果糖-1,6-二磷酸酶缺乏症
果糖-1,6-二磷酸酶缺乏症
果糖-1,6-二磷酸酶缺乏症属先天性常染色体隐性遗传。本症罕见。是由于肝脏缺乏果糖-1,6-二磷酸酶,以致自果糖转变为磷酸二羟丙酮与三磷酸甘油醛的生糖过程受阻,糖质异生主要来自乳酸与脂肪的糖酵解逆过程(见图)。因果糖-1,6-二磷酸酶缺乏,肝内有果糖-1,6-二磷酸、磷酸甘油及1-磷酸果糖沉积,导致磷酸化酶的抑制,从而使糖原分解障碍而产生低血糖、并有乳酸血症与丙酮酸血症。多数病婴的症状发生在生后几个月,饥饿或给予果糖饮食,即可出现低血糖症状,与果糖不耐受症不同,病婴一般没有呕吐。因脂肪肝或肝硬化而致肝肿大与肝功能损害。血乳酸浓度增高,当给予大量含果糖、氨基酸、甘油饮食或发生感染时,可产生乳酸性酸中毒。诊断靠以下特殊检查:
❶静脉果糖耐量(剂量同果糖不耐受症)试验,可出现低血糖与乳酸血症;
❷肝穿刺标本酶组织化学检查可证实果糖-1,6-二磷酸酶活性降低,从而获得确诊。治疗的关键是给予无果糖饮食,增加餐次以避免病婴饥饿,即可控制症状;低血糖时应静脉注射高渗葡萄糖。
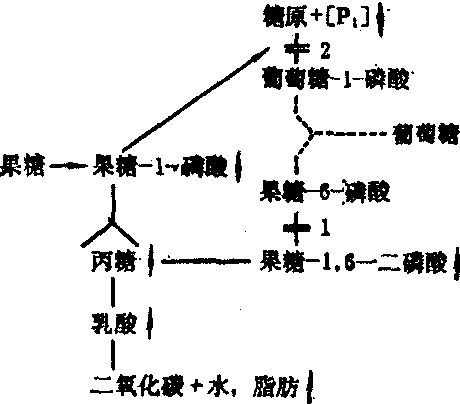
果糖-1,6-二磷酸酶缺乏
1 果糖-1,6-二磷酸酶缺乏
2 继发性磷酸化酶抑制
☚ 果糖不耐受症 粘多糖病 ☛
- 揭瓦亮椽是什么意思
- 揭瓴是什么意思
- 揭画皮是什么意思
- 揭疙儿是什么意思
- 揭疮疤是什么意思
- 揭痂巴是什么意思
- 揭痕是什么意思
- 揭白是什么意思
- 揭白传个神子是什么意思
- 揭白留真是什么意思
- 揭皇榜是什么意思
- 揭盅是什么意思
- 揭盒子是什么意思
- 揭盖是什么意思
- 揭盖儿是什么意思
- 揭盖头是什么意思
- 揭盖子是什么意思
- 揭短是什么意思
- 揭短儿是什么意思
- 揭短处是什么意思
- 揭短教唆是什么意思
- 揭短,说坏话是什么意思
- 揭石板集是什么意思
- 揭破是什么意思
- 揭碌碡是什么意思
- 揭碴是什么意思
- 揭 示是什么意思
- 揭示是什么意思
- 揭示的名单,指一个时期内的社会知名人士同登一榜是什么意思
- 揭示结局是什么意思
- 揭示结果或秘密是什么意思
- 揭示要旨是什么意思
- 揭示要领是什么意思
- 揭示(打字一)皓是什么意思
- 揭示,标明是什么意思
- 揭示,表明是什么意思
- 揭示,阐明是什么意思
- 揭祐民是什么意思
- 揭福是什么意思
- 揭秃疮是什么意思
- 揭秃疮嘎巴儿是什么意思
- 揭秃疮嘎渣儿是什么意思
- 揭秘是什么意思
- 揭穿是什么意思
- 揭穿别人动机的批评是什么意思
- 揭穿西洋镜是什么意思
- 揭穿隐讳的事是什么意思
- 揭穿,识破是什么意思
- 揭穿,说破是什么意思
- 揭竹蟀尾是什么意思
- 揭竿是什么意思
- 揭竿为旗是什么意思
- 揭竿四起是什么意思
- 揭竿斩木是什么意思
- 揭竿而起是什么意思
- 揭竿起事是什么意思
- 揭箧是什么意思
- 揭箧担囊是什么意思
- 揭箧探囊是什么意思
- 揭箸遮目是什么意思