红细胞破坏过多
在生理情况下,红细胞平均生存期约为120天。在其衰老过程中,红细胞主要在肝、脾中被破坏。红细胞正常生存期的维持,必须具备以下条件: 红细胞的膜、珠蛋白结构以及参与红细胞代谢的酶类必须正常; 血液中不存在破坏红细胞的异常凝集素或抗红细胞抗体; 单核巨噬细胞系统破坏红细胞的功能正常。红细胞破坏过多可发生于如下环节。
1. 红细胞膜的异常:人类红细胞膜是由蛋白质 (糖蛋白、脂蛋白)、脂质(磷脂、胆固醇、糖脂质)及无机离子所组成。蛋白质占49.2%,脂质占43.6%。膜蛋白中有膜收缩蛋白、膜动蛋白、各种酶(如磷酸甘油醛脱氢酶、6-磷酸葡萄糖脱氢酶(G6PD)、蛋白激酶、ATP酶、核苷酸代谢酶类等)。膜脂质中的胆固醇与磷脂之比约为3:4,二者占脂质的70~80%。红细胞通过膜收缩蛋白和膜动蛋白保持其正常形态及其变形性。通过其双层脂质、ATP酶、离子转运载体和钠泵,调节红细胞内外的物质交换,使红细胞内保持高钾、低钠和低钙。此外,红细胞膜上还有许多受体,能与某些物质特异地结合而起反应,如病毒受体,γ球蛋白、补体、某些激素的受体等。膜上的糖类有的与血型及受体的功能有关。
红细胞膜的异常现知有以下几种:
(1) 膜蛋白异常: 正常在ATP、蛋白激酶的作用下,膜收缩蛋白能进行磷酸化,然后与膜动蛋白结合成一种复合物。在病理情况下,膜收缩蛋白磷酸化若发生障碍,则膜的变形性受到限制,从而使膜变得脆弱,易受机械性损伤,以致红细胞可丢失部分膜而变成球形。此外,有人认为在上述情况下,钠易于进入红细胞内而钾则易漏出红细胞外。由于水随钠进入红细胞内,故红细胞更易变成球形。这种失去变形性的球形红细胞易在脾内阻留而遭破坏。遗传性球形红细胞增多症的发病机制可能与此有关。膜蛋白异常的另一种表现是蛋白组成异常。有人认为阵发性睡眠性血红蛋白尿的发病机制可能是获得性的膜蛋白组成异常。据研究,这种异常膜蛋白系糖蛋白,在其多肽链中缺乏含硫部分,缺陷部分可能是正常补体抑制所在部位,故这种红细胞对补体特别敏感。严重者红细胞对补体溶解作用的敏感性可达正常红细胞的20~30倍,故易被补体破坏而溶血。
(2) 膜脂质异常: 主要表现为膜上的胆固醇、磷脂异常。当红细胞膜上的胆固醇与磷脂之比明显升高时 (正常为3:4),膜易发生棘状突出。红细胞膜上发生以上改变的原因之一是缺乏卵磷脂-胆固醇酰基转移酶(LCAT)。LCAT活性减低时,血中胆固醇因不能酯化而增高,进入红细胞膜的胆固醇也增高,红细胞变成靶形。若这种改变比较严重,则当红细胞通过脾脏时,红细胞易变成有角刺状的棘细胞,这种细胞易被破坏而致溶血。若红细胞膜上的胆固醇和磷脂都增高,则易产生靶形红细胞,可见于某些肝病。先天性无β脂蛋白血症、酒精性肝硬变时也可见棘细胞增多,其机制也可能与膜上胆固醇、磷脂改变有关。
2. 红细胞酶的异常: 正常红细胞的葡萄糖代谢85~90%是通过糖酵解途径进行的,所产生的ATP是红细胞的主要能源。已知糖酵解过程中至少有10种酶可以发生遗传性缺乏。其中与产生溶血性贫血有关的酶是己糖激酶、葡萄糖磷酸异构酶、磷酸果糖激酶、二磷酸果糖醛缩酶、磷酸丙糖异构酶、二磷酸甘油酸变位酶、磷酸甘油酸激酶和丙酮酸激酶。其中以丙酮酸激酶缺乏症的发生率最高,占糖酵解途径中酶缺乏症的95%。以上各酶缺乏时,ATP产生减少,红细胞膜不能保持其正常的结构和功能,膜收缩蛋白的磷酸化、ATP酶、红细胞与血浆之间进行物质交换等功能不能正常地进行,红细胞的变形性受到影响,因而易被破坏而致生存期缩短。
正常红细胞的葡萄糖有10~15%是通过磷酸戊糖旁路进行代谢的,所产生的还原型辅酶Ⅱ(NADPH)使氧化型谷胱甘肽(GSSG)还原成为还原型谷胱甘肽(GSH),同时也能使高铁血红蛋白还原。在磷酸戊糖旁路中,现知与溶血性贫血有关的酶有六种,即6-磷酸葡萄糖脱氢酶、6-磷酸葡萄糖酸脱氢酶、谷胱甘肽还原酶、谷胱甘肽过氧化酶、谷胱甘肽合成酶和γ-谷酰半胱氨酸合成酶。其次,与此旁路有关的核苷酸代谢酶类如腺苷酸激酶等缺乏也可引起溶血。上述酶缺乏症中,以G6PD缺乏症最为常见。缺乏G6PD时,NADPH的生成减少,谷胱甘肽还原发生障碍。当患者服用某些氧化性药物(某些抗疟药、止痛退热药、磺胺类、砜类药等)、食物(如蚕豆)或发生感染时,红细胞内形成过氧化物(H2O2),使GSH氧化,所生成的GSSG由于NADPH减少,不能及时还原为GSH,故GSH进一步减少。正常红细胞中的GSH有保护血红蛋白上SH基的作用。由于GSH减少,生成的H2O2首先将珠蛋白β链93位半胱氨酸的SH基氧化,形成二硫键(蛋白-S-S-),血红素从血红蛋白上脱落,珠蛋白的肽链被拆开,使更多的SH基被暴露而氧化,血红蛋白乃发生不可逆性变化,形成变性珠蛋白小体(Heinz小体)。此种小体粘附在红细胞膜上,红细胞变形性降低,通过脾脏时,被阻留和破坏。3. 血红蛋白异常: 血红蛋白是由血红素与珠蛋白所构成。后者有二对肽链。HbA为α2β2,HbA2为α2δ2,HbF为α2γ2。α链有141个氨基酸,β链有146个氨基酸,δ与β链有10个氨基酸不同,γ与β链有39个氨基酸不同。每条肽链上有一分子血红素,其氨基酸的排列40%为直链,60%呈α螺旋型的二级结构,其本身又盘曲成立体空间的三级结构,成球形,中间的空腔为血红素。在整个血红蛋白分子中,四条肽链密切结合,形成四级结构。α1与β1,α2与β2各有34个连接点; α1与β2,α2与β1各有19个连接点。血红蛋白要保持其正常结构和功能,必须具备以下条件: 各肽链的氨基酸组成和排列正常,α与β链的各连接点稳定,分子的表面结构完整,带有正常电荷。
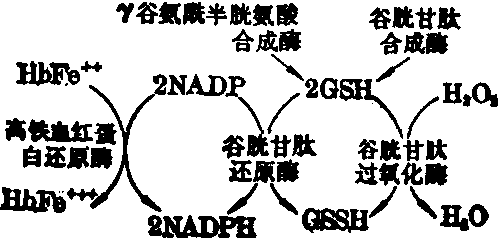
NADPH在还原谷胱甘肽、高铁血红蛋白中的作用
血红蛋白异常可表现为珠蛋白肽链的合成量异常。例如当α链合成障碍而都由β链所替代时,就形成4条β链的HbH,若都由γ链所替代,则可形成由4条γ链组成的Hb Bart。异常血红蛋白可表现为肽链中氨基酸被置换(如HbS是β链第6位谷氨酸被缬氨酸所置换),或肽链中氨基酸残基增多或缺失(如Hb Constant Spring为α链上增多31个氨基酸,又如Hb Gun Hill在β链的93~97位缺失5个氨基酸)。由于珠蛋白结构异常,故血红蛋白的功能可有改变。有的对氧的亲和力减低,氧合血红蛋白减少,引起发绀,如HbM Boston,HbM上海1,HbM上海2等; 有的对氧的亲和力异常增高,造成组织相对缺氧,促红素产生增多,引起红细胞增多,如Hb Hiroshima等。据1978年统计,异常血红蛋白已达320种以上,其中约半数表现为有一定程度的血红蛋白分子功能异常,其中部分异常血红蛋白可使红细胞易被破坏,导致溶血性贫血。其机制主要是异常血红蛋白不稳定或易聚合。不稳定血红蛋白如Hb Zurich,Hb GunHill等容易在红细胞内离解,进而形成包涵体,并沉积在红细胞膜上,使其变形性降低,易在脾内破坏; 异常血红蛋白如易聚合,则红细胞容易丢失膜而变成球形红细胞。例如在缺氧条件下,HbS变成一种螺旋状的聚合物,使红细胞变为镰刀形。这种细胞再与氧接触时,红细胞就丢失一部分膜而变成小球形红细胞。HbC在缺氧时聚合形成的一种副结晶,可粘附在红细胞膜上,当这种红细胞通过脾脏时,容易丢失部分膜而形成小球形细胞。
4. 血循环中存在异常抗体: 根据来源和性质,异常抗体可分为以下几种:
❶血型抗体。天然的如ABO抗体,这种抗体大多为完全性抗体,属IgG。获得性者如抗Rh抗体,这种抗体大多为不完全性抗体,多数属IgG或IgA。
❷药物抗体。如奎尼丁、大剂量青霉素所产生的抗体。大多为IgM,也可以是IgG。
❸自身免疫性抗体。可分为温抗体和冷抗体。温抗体都属不完全性抗体,多数为IgG,少数为IgM和IgA。可为原发的,也可继发于病毒感染、结缔组织病、慢性活动性肝炎、淋巴系统恶性增生性疾病(如慢性淋巴细胞白血病)或某些药物 (如甲基多巴、左旋多巴)的作用。冷抗体都是完全性抗体,在32℃以下时才起作用,可以是原发的(如原发性冷凝集素病),也可以是继发的(如继发于肺支原体病、传染性单核细胞增多症)。这类抗体属于IgM。阵发性冷性血红蛋白尿患者血清中存在一种冷热溶血素,这种溶血素在低温条件下附着在红细胞膜上,遇热时,溶血素发挥其作用而溶解红细胞。它是一种7S IgG型抗体。
IgM型抗体对红细胞的破坏作用特别强,红细胞膜上只要有20个IgM分子,即可与C1结合,使C4、C2及其他补体成分相继被激活,至C9被激活后就可直接溶解红细胞。此外,有IgM附着并有C3b结合的红细胞可被肝脏中的巨噬细胞破坏,因后者有C3b受体。有IgG型抗体附着的红细胞可不通过补体而附着在吞噬细胞上,继而在丢失部分细胞膜和膜的脂质后变成球形,最后在脾内破坏。IgG型抗体也可通过补体引起溶血,即抗体与抗原作用时,IgG的重链恒定区发生改变而结合C1q,补体系统乃被激活而破坏红细胞,但此时IgG型抗体需量很多。
5. 红细胞机械性损伤: 当微血管中因播散性纤维蛋白沉积而形成纤维蛋白网时,强行通过网眼的红细胞就会受到撕拉、挤压、扭曲,最后发生破裂(见于播散性血管内凝血、溶血尿毒症综合征)。此外,另有一种机制是当血流经过异常表面时,可因血流动力学改变而造成极强的旋涡。如果此时切变应力超过3,000dyn/cm2,就可破坏红细胞。这种情况见于心瓣膜病、人工瓣膜和心内修补术后。
6. 单核巨噬细胞系统破坏红细胞过多: 见于各种原因所引起的脾肿大伴脾功能亢进。
- (四)化肥试验是什么意思
- (四) 北京市农业环境监测站是什么意思
- (四) 北京市农工商联合总公司职工大学是什么意思
- (四) 北京市林业局是什么意思
- (四) 北京市水产科学研究所是什么意思
- (四) 北京解放后到改革开放前郊区基层党的建设是什么意思
- (四) 北部山地丘陵生态农业区是什么意思
- (四)区域差异大,发展不平衡是什么意思
- (四)区域开发研究是什么意思
- (四) 区域经济和企业规模逐步扩大是什么意思
- (四) 区域综合开发研究是什么意思
- (四)医学科技与医学教育是什么意思
- (四)医疗技术是什么意思
- (四)千里边疆文化长廊建设是什么意思
- (四)半殖民地半封建社会里的上海金融市场是什么意思
- (四)半粗毛羊生产基地是什么意思
- (四)华新禽蛋公司是什么意思
- (四)协调发展阶段是什么意思
- (四) 卖青是什么意思
- (四) 卫生是什么意思
- (四) 卫生事业是什么意思
- (四) 卫生体育事业是什么意思
- (四) 卫生医疗事业是什么意思
- (四)厅属事业单位是什么意思
- (四)厅属副厅级单位是什么意思
- (四)历史上有记载的最严重的干旱年是什么意思
- (四)历史事件是什么意思
- (四)历史沿革是什么意思
- (四)厦门华侨亚热带植物引种园是什么意思
- (四)厨房工程正在兴起是什么意思
- (四)县(市、区)水产技术推广站是什么意思
- (四)县级财政收入增加是什么意思
- (四)参加全国农民运动会是什么意思
- (四)及时验收,是什么意思
- (四)发展一批巩固一批是什么意思
- (四)发展以矿产资源开发为主的乡镇企业是什么意思
- (四)发展优势产业和拳头产品是什么意思
- (四)发展再生稻是什么意思
- (四)发展农场经济的主要措施是什么意思
- (四)发展农村经济的主要措施是什么意思
- (四) 发展壮大集体经济是什么意思
- (四)发展庭院经济,开辟致富财路是什么意思
- (四)发展提高时期是什么意思
- (四)发展提高阶段是什么意思
- (四)发展新党员是什么意思
- (四)发展新时期是什么意思
- (四)发展新阶段是什么意思
- (四) 发展水利经济是什么意思
- (四) 发展科技教育事业是什么意思
- (四)发展阶段是什么意思
- (四)发展集体经济,加强集体资产管理是什么意思
- (四)发挥了公路设施的载体功能是什么意思
- (四)发挥区位优势,发展特色乡镇企业是什么意思
- (四)发挥区域特点,大力兴办乡镇企业是什么意思
- (四)发挥小区优势,开发橡胶产业是什么意思
- (四)发放牧业贷款是什么意思
- (四)发菜是什么意思
- (四)口粮田加责任田是什么意思
- (四)古代加工的农产品是什么意思
- (四) 古墓葬是什么意思