永存共同动脉干
自心室发出的单一动脉干畸形,并由它分出冠状动脉、肺动脉及体动脉分支,称永存共同动脉干。系胎儿期干球嵴及肺动脉圆锥部不发育,原始共同动脉干不能正常地分隔成升主动脉及肺动脉所致。通常于动脉干下遗留巨大的室间隔缺损。
根据肺动脉的起源及部位,共同动脉干可分为四型(图):Ⅰ型,共同动脉干于其根部平行地分出单一的肺动脉干及升主动脉; Ⅱ型,左、右肺动脉起自共同动脉干的后壁;Ⅲ型,一侧或双侧肺动脉分别起自共同动脉干的侧壁;Ⅳ型,肺动脉及动脉导管均缺如,肺循环由支气管动脉或发自降、腹主动脉的分支供血。生前Ⅳ型几不能与伴有室间隔缺损的肺动脉闭锁即假性共同动脉干鉴别。共同动脉干与左、右心室通连,体、肺循环同时接受左、右心室的混合血,动脉血氧不饱和。肺循环承受体循环的压力,肺血流量增多,左、右心室负荷均增大,心室腔肥厚、扩张,终致心力衰竭。如肺血管阻力明显升高,更加重右心负荷形成Eisenmenger综合征。临床上病儿有不同程度的紫绀、呼吸困难及发育延迟,并常有心衰,病儿多在生后六个月内夭亡。胸骨左缘3~4肋间可闻及粗厉的收缩期杂音或伴有震颤。
平片表现:
❶心影多呈中度以上增大,以心室为主,可呈“斜卵形”拟似完全性大动脉转位。Ⅳ型者与重症心脏四联症无异;
❷“升主动脉”显著增宽,几为正常升主动脉的两倍,搏动增强。约1/3的病人合并右位主动脉弓;
❸两侧或单侧肺血增多,有肺门舞蹈及肺动脉高压的表现。1型者左肺门可高达主动脉弓水平呈“逗点”征,是有诊断意义的征象。Ⅳ型两肺门无肺动脉干支影,代之以细而乱的侧支循环;
❹与其他复杂畸形不同,其心脏及内脏位置多无异常。
造影表现: 心室或(和)升主动脉造影是唯一可靠的诊断方法。主要征象:
❶单一粗大的动脉干跨居于两心室之上,合并高位室间隔缺损,肺动脉分出后其管径迅速变窄。动脉干根部仅见一组半月瓣,瓣叶为2~4个不等,干瓣与二尖瓣前瓣纤维连接正常。
❷冠状动脉,一侧或双侧肺动脉、体动脉均起自此动脉干。Ⅳ型与假性共同动脉干的造影所见相同。
❸约1/3的病人可合并右位主动脉弓畸形。
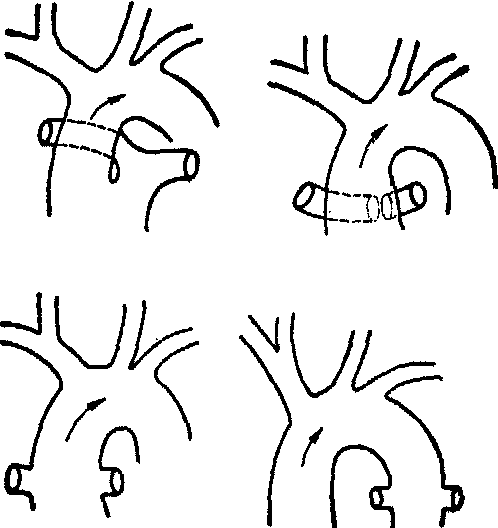
永存共同动脉干分型
左上Ⅰ型 右上Ⅱ型
左下Ⅲ型 右下Ⅳ型
- 杨鸿飞是什么意思
- 杨鹏是什么意思
- 杨鹏儒是什么意思
- 杨鹏升是什么意思
- 杨鹗是什么意思
- 杨鹤是什么意思
- 杨鹤是什么意思
- 杨鹤是什么意思
- 杨鹤书是什么意思
- 杨鹤侪是什么意思
- 杨鹤龄是什么意思
- 杨鹤龄是什么意思
- 杨鹤龄是什么意思
- 杨鹤龄是什么意思
- 杨黄美幸是什么意思
- 杨黄门奏疏是什么意思
- 杨黼是什么意思
- 杨鼎勋是什么意思
- 杨鼎勋是什么意思
- 杨鼎川是什么意思
- 杨鼐是什么意思
- 杨齐福是什么意思
- 杨龙九是什么意思
- 杨龙生是什么意思
- 杨龙生是什么意思
- 杨,亨是什么意思
- 杨,约是什么意思
- 杩槎是什么意思
- 杪秋寻远山,山远行不近。是什么意思
- 杭是什么意思
- 杭是什么意思
- 杭·杜亚传是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏是什么意思
- 杭世骏(1695—1772或1696—1773)是什么意思
- 杭丽制冷设备有限公司是什么意思
- 杭人唐云是什么意思
- 杭勇琪是什么意思
- 杭县是什么意思
- 杭城摩擦材料有限公司是什么意思
- 杭子和是什么意思
- 杭子梢是什么意思
- 杭宁路是什么意思
- 杭州是什么意思
- 杭州是什么意思
- 杭州是什么意思
- 杭州是什么意思
- 杭州是什么意思
- 杭州是什么意思
- 杭州4日游(游程参考)是什么意思
- 杭州——富春江——千岛湖游是什么意思
- 杭州——海宁——嘉兴——湖州游是什么意思
- 杭州——黄山之旅是什么意思