毒物鉴定
毒物鉴定常遇到两种情况: 一种是已知毒物中毒的鉴定,要求确证,并测定该毒物或其代谢产物在组织和体液中的分布和含量,选用适宜的检材和分离方法,进行定性和定量测定;另一种情况是未知毒物的鉴定。引起中毒的毒物繁多,案情各异,故只能根据案情、中毒症状和尸体解剖所见,审慎地排除某些毒物中毒的可能性,参考各地区常见毒物中毒和死者生前用药情况,制定初步分析方案,使其既照顾到面,又有重点地分离某种或某几种最可能存在的毒物。
法医毒物鉴定一般可分为三个阶段:初步定性分析,毒物的鉴定,毒物的定量。初步定性分析所采用的方法应很灵敏和无损(不破坏或消耗毒物),如紫外吸收光谱、薄层色谱、免疫测定等。重点放在类反应方面,以确定某类毒物的存在与否。根据第一阶段初步定性分析提供的线索,修订分析方案,选用适宜的检材和可靠的方法,分离和纯化体液和组织中的毒物,做出确实、可靠的鉴定。但应注意不能仅依据一个或两个非特异性的反应,或一个溶剂系统的比移值(Rf)即确定是何种毒物。宜采用两种以上不同性质的鉴定方法,并用已知毒物做参比。有时还应作原植物、动物的鉴定及动物试验。最后测定其含量。
毒物初步定性分析 法医毒物鉴定中常用的几种初步定性分析方法和一些常见毒物的数据分述如下。
液上气体分析 检材(如体液等)置于密闭容器中在38℃保温,其上部气体可供气相色谱分析挥发性毒物。于三个容积为15ml具有异丁橡胶垫螺旋盖的密闭小瓶中,分别加1gNaCl。 一瓶加1ml检材(血或其它试样);一瓶加低浓度参比溶液; 另一瓶加高浓度参比溶液,盖严,旋摇混匀,在38±0.05℃水浴中保温45分钟。用预热到38℃的1ml结核菌素注射器,抽取液面上部气体0.25ml,立即注入气相色谱仪。依次分析低浓度参比溶液、检材和高浓度参比溶液上部气体,各记录色谱图5分钟。气相色谱柱长2m×3mm,填充Porapak S(80~100目),载气为氦气 (55psig 45ml/min)或氢气 (20psig45ml/min)或空气 (15psig 330ml/min),温度: 柱温为165℃,进样器温为175℃,火焰离子化鉴定器温为225℃。
尿的颜色反应 多种毒物或其代谢产物皆从尿中排出。故尿的颜色反应,系常采用的一种初步定性反应。可直接取尿检材作附表所列的反应。
铜片试验 检材在稀盐酸溶液中与铜箔一起加热。若有汞、砷、锑、铋等常见有毒金属存在,则铜箔上有沉淀物附着,可供进一步鉴定。
尿的颜色反应
| 毒 物 | 反 应 |
| 水杨酸 吩噻嗪 三氯化合物(水 合氯醛等) 丙咪嗪 | 0.5ml尿+3滴5%FeCl3 紫色 1滴尿+2滴FPN试剂 蓝紫 1m120%NaOH+1ml重蒸吡啶, 吡啶层 沸水浴加热2分钟+2ml尿加热红色 1ml尿+1ml Forrest试剂 绿色 (吩噻嗪 紫色,消退) |
| 对-氨基苯酚 (非那西汀等的 代谢物) 硫醚嗪 | 2ml尿+稀HCl酸化+3滴1% NaNO2+3滴新配1%α-萘酚的 10%NaOH溶液 红色 1ml尿+1mlFeCl3-H2SO4(5% FeCl3 1ml+10N H2SO4至50ml)红蓝 |
FPN试剂 5ml 15% FeCl3+45ml 20%(w/w)HClO4+
50ml 50% HNO3
Forrest试剂 25ml 0.2% K2Cr2O7+25ml 30% H2SO4+
25ml 20% HClO4+25ml 50% HNO3
取铜箔 (5×10mm),依次经硝酸(3:20)、水、乙醇、乙醚洗净,放锥形瓶中,加4ml浓HCl,20ml尿(或胃内容物或10g组织匀浆加10ml水)。加热煮沸1小时,蒸发过程损失的溶液,加盐酸(1:5)补足。取出铜箔洗净,如呈银白色提示有汞,色变暗可能含砷、锑、铋、硒、锌、硫。
免疫测定 一种药物如吗啡、巴比妥等作为半抗原与大分子载体如牛血清蛋白(BSA)或合成的多肽等结合成为完全抗原(Ag),加入佐剂,免疫动物,产生对药物具有特异性的抗体(Ab),分离免疫动物的抗血清中的抗体,即可用来测定该药物。采用药物的免疫测定法测定尿或血清中的药物,不需要经过分离提取,操作简便、快速、灵敏度高(微克至毫微克水平)。与蛋白质或葡萄糖醛酸结合的药物也可以检出。但具有相同结构的化合物,如巴比妥、苯巴比妥、异戊巴比妥等都呈阳性反应,不能鉴别。所以本法特别适宜于初步鉴定或大量检材的自动分析,本法也可用于毒物定量。本法作为阴性排除试验尤为简便。
常用的药物免疫测定法有:
❶放射免疫测定(RIA)。
❷酶抑制免疫测定(EMIT)。
❸血凝抑制免疫测定(HI)。
❹自由基测定(FRAT)。
RIA、EMIT、HI和FRAT方法的比较
| 方 法 | 优 点 | 缺 点 |
| 放射免疫 (RIA) | (1)灵敏度(μg/ml) 吗啡0.03~0.06 巴比妥类0.05~0.10 苯丙胺0.10 (2)判断客观 | (1)需时1~2h (2)使用放射性同位素 (3)仪器和试剂贵 |
| 酶抑制免 疫测定 (EMIT) | (1)操作简便、快速 (2~4min) (2)判断客观 | (1)如尿中有溶菌酶呈 假阳性,须做空白 对照 |
(续上表)
| 方 法 | 优 点 | 缺 点 |
| (3)灵敏度(μg/ml) | (2)灵敏度低于其它免 疫测定法 | |
| 吗啡0.5 巴比妥类1.5~2.0 苯丙胺2.0~2.5 可卡因1.0~1.5 美散痛0.5 | (3)试剂贵 | |
| 血凝抑制 免疫测定 (HI) | (1)操作和设备简单 (2)灵敏度可调节 (μg/ml) 吗啡0.03~0.06 可卡因0.03~0.06 美散痛0.03~0.06 | (1)需时1.5~2h (2)判断主观 (3)尿沉淀判断困难 (4)试剂质量不一致 |
| 自由基测 定 (FRAT) | (1)操作快速(2~4min) (2)灵敏度(μg/ml) 吗啡0.5 巴比妥类1.0~2.0 苯丙胺1.0~2.0 可卡因0.5~1.0 美散痛0.5~1.0 | (1)需要贵重仪器 |
毒物鉴定及定量测定 初步定性分析所获得的结果,若与案件情况、中毒临床症状和尸体解剖材料无矛盾,表明有某一类毒物存在,即应采用适当的分离方法,分离出毒物并纯化,进行毒物鉴定及其含量测定。
目前毒物鉴定和定量测定的方法,常用的有下述几种。若将气相色谱与质谱联用,能完全满足毒物鉴定所要求具备的灵敏、快速、精确、可靠等条件。
色谱法 混合物中的各组分,在某种流动相的影响下,在固定介质中具有不同的迁移速率。根据这一原理,可将混合物中的各组分分离、分出。这类方法,称为色谱法。
色谱法于六十年代应用于法医毒物分析,起了极大的推动作用。薄层色谱、纸色谱、气相色谱和高压液相色谱法是效率极高的微量分离方法; 其中薄层(纸)色谱设备简单,操作简便,灵敏,快速,分离效果好,得到广泛应用。近年来,气相色谱也已成为广泛采用的技术。过去,法医毒物分析的最大困难之一,是从含有大量杂质的内脏组织中分离微量(毫克以至微克水平)的有机毒物。一百多年来,Stas-Otto以及许多改良的方法,用有机溶剂提取分离,只能得到粗提物,都不能解决微量毒物的纯化,使毒物难以得到可靠的证明。色谱法不仅使毒物和杂质分离,使碱性毒物和腐败尸体产生的碱性物质分离,还可以使毒物和其结构近似的代谢物得到分离。经色谱法分离的毒物便可采用红外吸收光谱和质谱分析作出可靠的鉴定。
薄层色谱法(TLC)和纸色谱法(PC): 薄层色谱法比纸色谱法快速(TLC仅需几十分钟,PC需几小时至几十小时),灵敏度高(TLC点样1~10μg,有时可分离十分之几微克的试样,PC点样要几十至几百微克),分离效果好(TLC分离的色谱斑比PC清晰),TLC可用腐蚀性显色剂如浓硫酸等,所以薄层色谱法愈来愈广泛地被采用。但纸色谱负荷量大,分离含杂质较多的粗提物较好。在薄层(纸)色谱中,一个化合物显色后的色斑常用Rf(比移值)来表示。Rf=某一化合物色斑中心距起线的距离/溶剂前沿距起线的距离
在一定实验条件下,Rf值是一个化合物的重要的参数。薄层(纸)色谱采用Rf值和显色反应来识别一化合物。薄层(纸)色谱可以利用显色反应提示可能含有某类化合物,这点比气相色谱有利。但Rf值受许多实验条件的影响,如温度、吸附剂活化的条件、存放过程中吸水的量、混和溶剂在展开过程中组分的变化、溶剂的纯度、容器的大小及是否为溶剂蒸气所饱和等。所以书上所列Rf值仅供参考。鉴定时,必须取已知毒物在同一块板上展开与未知毒物比对,并选用适宜的溶剂系统,使毒物的Rf值在0.2~0.8之间。
应注意不能根据一种溶剂系统得到的一个Rf值来确证一毒物。必须选用两种以上不同溶剂系统来判别已知化合物和未知化合物的Rf值是否相同。当然,为了严格确证一个毒物,最好将薄层(纸)色谱分离纯化的未知毒物,选用其它测定方法如红外吸收光谱、质谱等来鉴定。
薄层板和展开溶剂的选择:常用的薄层板有硅胶、氧化铝和纤维素板。一般非极性药物选用非极性溶剂展开,极性较高的药物选用极性较高的溶剂展开。最常用的硅胶活性强,甚至毫克量试样都可用薄层色谱分离,硅胶为弱酸性,强碱性药物可能与它生成盐,停留于原点,故所用溶剂常加二乙胺或用N/10 NaOH调硅胶制薄层板。
色斑(色谱)的显示,色谱即样品中各组分呈现不同位置的几个色斑,可用物理的或化学显色方法来显示色斑。宜先用物理的无损方法,如在254nm波长紫外线下,多数生物碱皆有荧光,巴比妥酸盐类安眠药和多数芳香族化合物呈暗斑,可标出色斑位置。含有荧光物质F254的硅胶板,在254nm波长紫外线下,荧光背景上荧光淬灭的暗斑更为清晰。化学显色方法,可将薄层板溶剂挥散后,置于含碘蒸气的容器中薰之,有机毒物即显棕色斑,可取出标明位置。如系生物碱类采用碘化铋钾、碘铂酸盐;芳胺采用对-二甲氨基苯甲醛; 伯胺类采用茚三酮;酚类采用酚试剂;巴比妥酸盐类采用汞试剂和二苯偕肼腙等。下表列出一些药物的薄层色谱的Rf值。溶剂Ⅰ可用于初步预试验以探索可能存在的毒物。
气相色谱法: 主要指用气液填充色谱柱。色谱柱为玻璃和不锈钢制成,内径2~6mm,一般长1~4m ,填充以一种惰性物质(载体),载体表面涂一层非挥发性液体(为固定相),以不与样品起化学反应的气体 (载气) 为移动相。载气带着试样蒸气通过色谱柱,混合物各组分即不断地分配于移动相(气体)和固定相 (液体) 中而得到分离。分离开的各组分通过灵敏的鉴定器转换为电压或电流讯号显示出来。
一些药物薄层色谱的Rf值
| Rf×100 | ||||||
| Ⅰ | Ⅱ | Ⅲ | Ⅳ | Ⅴ | Ⅵ | 药 物 |
| 24 35 42 | 54 | 20 31 | 苯巴比妥 巴比妥 美多眠 | |||
| 56 59 | 75 | 58 66 | 异戊巴比妥 戊巴比妥 | |||
| 60 68 | 77 | 63 77 | 速可眠 安眠朋 | |||
| 33 34 63 69 | 59 | 眠尔通 苯妥英 凡眠特 美介眠 | ||||
| 75 25 35 55 60 | 81 38 | 导眠能 扑热息痛 安替比林 非那西丁 氨基比林 | ||||
| 26 | 16 | 57 | 奋乃静 | |||
| 45 52 | 55 55 | 36 47 | 65 78 | 丙嗪 异丙嗪 | ||
| 53 58 61 | 75 77 | 61 58 58 | 45 44 52 | 73 63 75 | 硫醚嗪 氯丙嗪 氟丙嗪 | |
| 5 | 14 | 0 | 28 | 24 | 吗啡 | |
| 6 | 14 | 11 | 6 | 阿托品 | ||
| 12 | 19 | 18 | 8 | 麻黄碱 | ||
| 14 15 17 | 32 | 7 54 20 | 47 | 50 | 奎宁 毒芹碱 马钱子碱 | |
| 17 20 20 26 | 35 39 | 27 16 | 17 28 18 | 29 39 | 士的宁 可待因 去氧麻黄碱 毛果芸香碱 | |
| 27 | 64 | 49 | 异烟酰异丙肼 | |||
| 36 | 54 | 67 | 东莨菪碱 | |||
| 36 | 45 | 35 | 28 | 苯丙胺 | ||
| 37 37 | 40 58 | 美散痛 苯海拉明 | ||||
| 39 | 55 | 81 | 咖啡碱 | |||
| 42 54 58 | 64 70 | 57 49 | 52 48 35 | 62 63 57 | 烟碱 度冷丁 丙咪嗪 | |
| 70 75 | 55 | 乌头碱 蛇根碱 | ||||
| 77 | 97 | 安眠酮 | ||||
| 78 | 84 | 73 45 | 57 | 73 74 | 可卡因 利眠灵 | |
| 74 | 65 | 94 | 安定 | |||
溶剂: Ⅰ 苯:二氧六环:丙酮:氨液(50:20:28:2)
Ⅱ 丙酮:氯仿:二氧六环:氨液(10:90:95:5)
Ⅲ 环己烷:氯仿:二乙胺(50:40:10)
Ⅳ 乙酸乙酯:正己烷:氨液(20:9:10)
Ⅴ 甲醇
Ⅵ 氯仿:甲醇(90:10)
薄层板: Ⅰ、Ⅱ、Ⅲ、Ⅳ和Ⅵ用硅胶G
Ⅴ,用0.1N NaOH处理的硅胶G
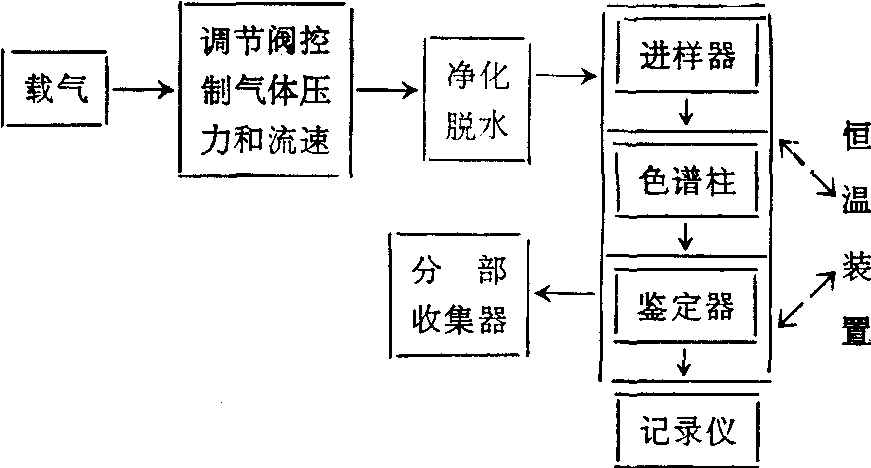
气相色谱示意图
气相色谱可分离在400℃以内不分解,具有0.2~10mm Hg蒸气压的物质,或分解为恒定产物的物质。如吗啡在254℃即部分分解,但在254℃以下已有足够蒸气可供分离检测。
保留时间(tr)和死时间(t°r): 保留时间(tr)表示试样各组分从色谱柱流出的先后,可做为鉴定各组分的定性指标。死时间(t°r)为载气进入色谱柱到出现浓度极大点的时间(OA)。保留时间(tr)为样品从进样开始到出现浓度极大点的时间 (如图中OB所示)。校正保留时间(t′r)为保留时间减去死时间,如图中AB所示。相对保留值(α)是以一种物质作为标准,而求出其他物质的保留值(t′ri)对此标准物(t′rs)的比值 i表示测定物,s表示标准物。
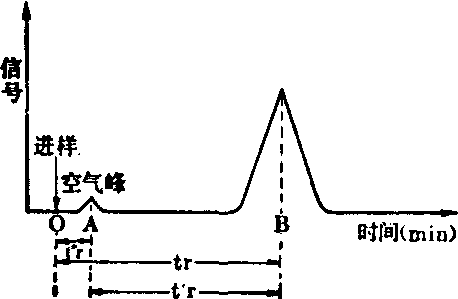
tr和t°r 示意图
根据色谱图试样峰面积与加入的精确称量的标准物峰面积的比,可求出试样的含量。气相色谱分离效率高(可分离结构相近的药物以及其代谢物),灵敏度高(微克以下),方法快速。气相色谱的缺点是它只记录下分离化合物的峰,唯一指标是各组分的保留时间而并不指明各组分的性质。保留时间和薄层色谱的Rf值一样,受许多实验条件影响。故判断未知物与已知物是否相同时,同样不能只根据一根色谱柱所测tr值一致,而应采用不同极性固定液的色谱柱测定二者的保留时间是否相同。并必须结合其它物理学或化学方法测得的结果,做最后判断。采用分部收集器,可分别收集不同的纯品,供进一步鉴定。
(1) 载气,常用的载气有氢气、氮气、氦气和氩气等。
(2) 载体,要求表面积大、多孔、孔径及分布较均匀,表面无或仅有很弱的催化或吸附性能,热稳定性及机械强度均较好。常用的载体为硅藻土制品:
❶101白色硅藻土载体(chromosorbw)。
❷201红色硅藻土载体(chromosorbP)。
❸102硅烷化白色载体(chromosorb AW HMDS)。
(3) 固定液,固定液的选择极为重要,是有效地分离混合物各组分的决定因素之一。固定液一般分为非极性、中等极性和极性三类。毒物分析常用的固定液有:
❶非极性固定液:硅橡胶(SE-30,OV-1)最常用作分离碱性药物,一般按分子量大小分离,最高操作温度250℃。“阿匹松”润滑脂L(APiezon L) 用作分离巴比妥酸盐类和中性药物。
❷中等极性固定液: 聚氯代烷基硅氧烷(QF-1即FS-1265),苯基硅氧烷(OV-17)。
❸极性固定液: 聚乙二醇(carbowax,PEG),硬脂酸酰胺(Hallcomid M-18)。分离挥发性毒物,现多采用甲基苯乙烯-二乙烯苯共聚物多孔微球(Porapak Q)。
(4) 柱的选择,毒物分析初步试验,应用少数色谱柱,可用2.5%SE-30或OV-1测碱性毒物,用10%“阿匹松”L测巴比妥酸盐和中性化合物,用Porapak Q或PEG400测挥发性毒物。文献所载实验条件仅供参考,应取标准物,选用适宜的流速,测得实际最大的柱效率。并采用能得到合理的分析时间的最低温度。
为使未知物实验数据能与文献资料查对,Kovats采用保留指数(Ix)
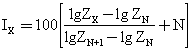 Z表示保留值如保留时间t′r,N表示正构烷烃的碳数,X表示待定的未知物。
Z表示保留值如保留时间t′r,N表示正构烷烃的碳数,X表示待定的未知物。被测物的校正保留值应在两个正构烷烃的校正保留值之间。Kazyak汇集了常见药物的保留指数,采用三种固定液(OV-1,OV-17,QF-1)的色谱柱。
Finkle采用气相色谱,4根色谱柱,3种固定液,用氯仿在中性和碱性条件下直接从检材(3ml血或尿,1g组织匀浆)提取毒物、药物及其代谢物,蒸发氯仿,溶于20μl乙醇:氯仿(1:1)中,取1μl进柱。测定了600种物质的相对保留时间。下表选录一些常见毒物的相对保留时间。
一些毒物的相对保留时间(系统A)
| 相对保留时间(min) | 药 物 |
| 0.19 0.23 0.23 0.24 0.28 0.38 0.42 0.55 0.58 0.80 0.91 1.00 1.09 1.11 | 水杨酸 乙酰苯胺 凡眠特 美解眠 华法灵 巴比妥 可拉明 异烟肼 异烟酰异丙肼 非那西丁 异戊巴比妥 戊巴比妥 五氯苯酚 度冷丁 |
(续上表)
| 相对保留时间(min) | 药 物 |
| 1.18 1.21 1.30 1.36 1.50 1.65 1.68 1.75 1.92 2.00 2.20 2.36 | 眠尔通 速可眠 咖啡因 毒扁豆碱 导眠能 硫喷妥 苯妥英 氨基比林 洋地黄毒苷 马拉硫磷 对硫磷 苯巴比妥 |
U形玻管,2m×4mm内径,80/100目。chromosorbG涂以2.5%SE-30。柱温约200℃,调节至苯巴比妥标准保留时间为3.5±0.2min。进样器:240℃。FID鉴定器220℃。氮气流速:约40ml/min。
一些毒物的相对保留时间(系统B)
| 相对保留时间(min) | 药 物 |
| 0.13 0.17 0.20 0.31 0.42 0.46 0.53 0.53 0.54 0.54 0.60 0.61 0.61 0.72 0.82 0.86 0.88 0.94 1.00 1.26 1.40 1.43 1.51 1.75 | 度冷丁 丙胺卡因 利多卡因 普鲁卡因 安眠酮 美散痛 莨菪碱 山道年 阿托品 阿卡因 DDT 丙米嗪 氟丙嗪 异丙嗪 东莨菪碱 丙嗪 去甲羟安定 保泰松 可待因 安定 吗啡 苯妥英 氯丙嗪 利眠宁 |
U形玻管,60cm×4mm内径,80/100目。Chromosorb G涂以2.5%SE-30。柱温约200℃,调节至可待因的保留时间为3.8±0.2min。进样器:240℃。鉴定器:220℃。氮气流速:约40ml/min。
光谱法 光是一种电磁波,具有波动性和微粒性,人眼可感觉到的光,波长在380~780nm之间,称可见光区。波长为200~400nm的称为紫外线;波长为780~1000nm的称为近红外线,波长再长者为远红外线,最长可达5mm。光波从紫外到红外排列,称为光谱。利用光谱学的原理和方法,藉以确定物质的结构及其组分的一类分析方法,称为光谱法。光谱分析法在分析化学中占有重要位置。既可定性,又可定量,为一类有效、快速、微量、准确的分析测定方法,适合于法化学的要求。光谱分为发射光谱和吸收光谱。
紫外吸收光谱法: 紫外吸收光谱是由不同分子结构的物质,对紫外光区电磁辐射选择性吸收所产生的分子吸收光谱。它是由于多原子分子的价电子发生跃迁,如双键π电子和未共享电子对吸收一定频率辐射能,从基态跃迁至较高能级的激发态而产生。所以紫外吸收光谱主要决定于共轭双键结构以及共轭双键与有不成键电子对的基团连结的情况。一般紫外分光光度计测定的波长范围为200~400nm。
吸收光谱即以波长为横座标,吸光度(A)为纵座标,标绘一个化合物对不同波长辐射的吸收曲线图。
吸收峰的波长最大称吸收值以λmax表示。溶剂和pH对一些化合物的吸收光谱影响大,如巴比妥在pH10时λmax为240nm,在pH13时为255nm,在pH2时吸收峰消失。
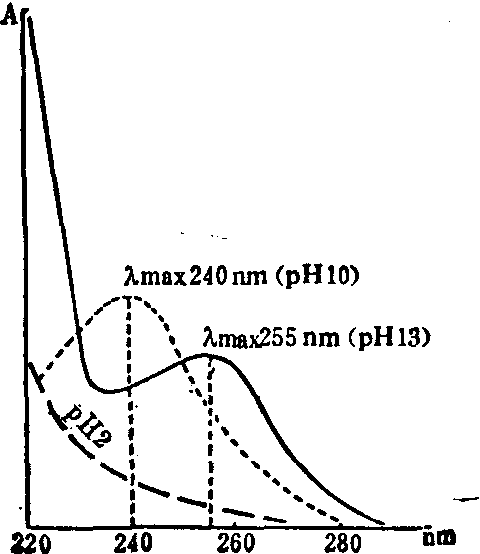
5,5-取代巴比妥酸盐的吸收光谱

5,5-取代巴比妥酸盐
由于C5上的取代基R1和R2与母环不形成共轭,对吸收光谱无影响,所以巴比妥、异戊巴比妥、速可眠等同属5,5-取代巴比妥酸盐类安眠药,为同一族化合物,它们都有相同的吸收光谱,在pH10时λmax都等于240nm,在pH13时的λmax都是255。因此用紫外吸收光谱不能鉴别这些药物。
λmax是一个药物或一类型药物的重要参数,为紫外吸收光谱鉴定一化合物的重要依据。由于仪器和实验条件的差异,资料记载的λmax值可能相差2~4nm。因此鉴定未知毒物时应同时检测已知毒物的吸收光谱。E1%cm为一化合物浓度为1%,光径为1cm时在λmax的比吸光系数。
 为克分子吸光系数,即1克分子浓度的某一化合物,吸收层厚度为1cm时的吸光度。一个化合物的溶液由于pH改变而结构改变。吸收峰向长波长一端移动称为向红效应。向短波长移动称为向紫效应。观察在酸性和碱性介质中吸收峰的移动有助于毒物的鉴定,如碱性毒物吗啡,溶于N/10H2SO4测定吸收光谱λmax为284nm,加碱变碱性后λmax为297nm。
为克分子吸光系数,即1克分子浓度的某一化合物,吸收层厚度为1cm时的吸光度。一个化合物的溶液由于pH改变而结构改变。吸收峰向长波长一端移动称为向红效应。向短波长移动称为向紫效应。观察在酸性和碱性介质中吸收峰的移动有助于毒物的鉴定,如碱性毒物吗啡,溶于N/10H2SO4测定吸收光谱λmax为284nm,加碱变碱性后λmax为297nm。常见药物紫外吸收光谱的吸收峰
| 吸收峰 λmax (nm) | E1%1cm | 溶 剂 | 在强碱性溶 液中λmax (nm) | 药物 |
| 240 | 310~ 550 | pH10硼酸 盐缓冲液 | 255(pH13) | 5,5-取代巴 比妥酸盐 (如巴比妥, 苯巴比妥, 异戊巴比 妥,速可眠 等)pH2吸 收峰消失 |
| 250 | pH10硼酸 盐缓冲液 | 240(pH13) | 1,5,5-取代 巴比妥酸盐 (如安眠朋) | |
| 305,255 | pH10硼酸 盐缓冲液 | 305、250 (pH13) | 硫巴比妥酸 盐(如硫喷 妥钠) | |
| 235 | 880 | N/2 NaOH | 迅速水解吸 光度降低 | 导眠能 |
| 251,257 | 乙醇 | 导眠能 | ||
| 250 229 230 242 244 256 300 | N/10NaOH N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/2 H2SO4 | 同上 | 美介眠 阿斯匹林 安替比林 扑热息痛 非那西丁 氨基比林 水杨酸 | |
| 249,300 252,300 254,306 | N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 | 253 257 258 | 异丙嗪 丙嗪 奋乃静 | |
| 255,307 256 263,230,313 228,272,279 233,275 | 1280 | N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 | 258 258 275 285 270 | 氯丙嗪 三氟拉嗪 硫醚嗪 普鲁卡因 可卡因 |
| 234,269 234,275 | 1280 | N/10 H2SO4 N/10 H2SO4 | 264 | 安眠酮 乌头碱 |
| 241,284,359 245,306 | 1402 | N/10 H2SO4 N/10 H2SO4 | 250 260 | 安定 利眠宁 |
| 250,316,346 251 253,259 | 1005 | N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 | 280 | 奎宁 丙咪嗪 美散痛 |
| 255 260 265 267 | 315 | N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 N/10 H2SO4 | 士的宁 烟碱 马钱子碱 异烟酰异丙 肼 | |
| 272 | 470 | N/10 H2SO4 | 咖啡因 | |
| 284 284 | 43 | N/10 H2SO4 N/10 H2SO4 | 297 | 吗啡 可待因 |
紫外分光光度法不专一,同类型药物常有相同的吸收光谱,它不能鉴别长效与短效巴比妥类安眠药,它不能区别苯并二氮䓬类药物,安定与去甲羟基安定,不能分辨一些药物及其代谢物。它的灵敏度也不很高,有些药物浓度在0.2mg/100ml以下不能检测。
但作为一种无损的分析方法,操作简便,设备易于得到,急性中毒时药物浓度一般较高,一些有机毒物皆有可资鉴识的吸收光谱,因此可做为初步鉴定或与其他如色谱法等合用于毒物鉴定。
红外吸收光谱法: 每一种有机化合物皆有代表其独特的物理性质的红外吸收光谱。除光学异构体外,尚没有两种化合物具有相同的吸收曲线。红外吸收光谱法除用于定性分析,也可用于定量分析。但定性分析上更重要。一般红外分光光度计测定的范围为2~25μm,常用波数(
 )为单位,即每厘米长度中的波数,波数为波长的倒数,单位为cm-1。2~25μm相当于5000~400cm-1,属中红外部分,简称红外。
)为单位,即每厘米长度中的波数,波数为波长的倒数,单位为cm-1。2~25μm相当于5000~400cm-1,属中红外部分,简称红外。当一个化合物通过红外辐射,电子能级即从基态跃迁于旋转能级和振动级之间,乃产生化合物特征性的吸收光谱。红外吸收光谱中,波数在1400cm-1以上的吸收峰属于功能团或键的振动作用所引起,如含苯环的化合物在1500cm-1附近都有一个吸收峰。波数在1400cm-1以下的吸收峰大多属于整个分子的振动作用。1600~650cm-1区段吸收带复杂,来源尚未弄清楚,称为“指纹”区。
供红外吸收光谱测定的样品必须是纯品。经薄层(纸)色谱或气相色谱纯化的毒物可供红外分析用。
毒物经溶剂提取分为酸性、中性和碱性部分毒物,再经薄层、气相色谱分析推断可能是何毒物,分出经色谱法纯化后的毒物,即可采用红外光谱分析进行鉴定。
未知毒物红外吸收光谱的鉴定,可根据Sadtler方法检索。参看下图和说明。
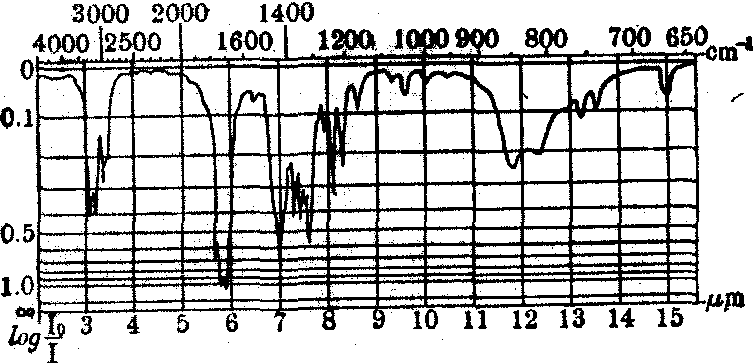
异戊巴比妥的红外吸收光谱
将上图红外吸收光谱每一微米区间的最强吸收带波长登记在下图Sadtler编码卡上。
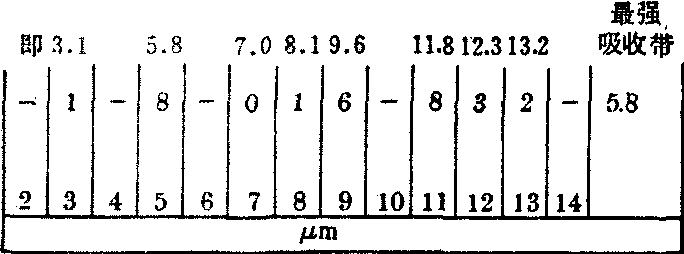
Sadtler异戊巴比妥红外吸收光谱编码卡
(1)在下列记录卡上登记未知毒物红外吸收光谱每1μm区段中最强的吸收带至近0.1μm。如7~8μm区间最强吸收带为7.0μm,8至9区间最强吸收带为8.1,即在卡片上7μm处写上0,在8μm处写上1。
(2)在整个吸收光谱中选出最强的吸收带,在图中为5.8,即在卡片上最强吸收带一项记上5.8。
(3)在Sadtler的药物红外吸收光谱检索表中,查到最强吸收带5.8 (该表系按最强吸收带波长顺序排列)。然后,比对各微米区间的数字 (允许可能相差±0.1或0.2),如符合,即根据序号(如R33))查到吸收光谱图为异戊巴比妥,仔细对比,如相符即可鉴定为该未知毒物为异戊巴比妥。几种巴比妥酸盐红外吸收光谱编码见下表。
几种巴比妥酸盐类安眠药的红外吸收光谱编码
| μm | 最强吸 收带 | 检索 号 | 药 物 | ||||||||||||
| 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |||
| 1 | 8 | 3 | 1 | 58 | R740 | 戊稀巴比妥 | |||||||||
| 1 | 8 | 0 | 2 | 4 | 6 | 9 | 58 | R317 | 环己烯巴比妥 | ||||||
| 1 | 8 | 0 | 1 | 6 | 8 | 3 | 2 | 58 | R33 | 异戊巴比妥 | |||||
| 1 | 8 | 0 | 2 | 6 | 8 | 2 | 4 | 58 | R480 | 戊巴比妥 | |||||
| 2 | 8 | 9 | 2 | 0 | 6 | 6 | 4 | 4 | 4 | 58 | R65 | 巴比妥 | |||
| 2 | 8 | 9 | 3 | 3 | 5 | 6 | 1 | 2 | 1 | 58 | R410 | 甲黑西塔耳 | |||
| 1 | 8 | 0 | 3 | 3 | 3 | 3 | 5 | 3 | 5 | 1 | 58 | R403 | 甲巴比妥 | ||
| 2 | 9 | 4 | 2 | 0 | 4 | 59 | R492 | 苯巴比妥 | |||||||
| 1 | 9 | 5 | 4 | 5 | 8 | 0 | 2 | 6 | 59 | R690 | 硫喷妥 | ||||
质谱法 是将有机化合物转变为快速运动的带正电的分子离子和碎片离子,然后再进行分离和鉴定的方法。对不同电离子的分辨,主要是根据其在磁场或静电场中,或者这二种场都存在时,正电离子径迹的不同。部分也根据在无场区域离子运动速度的差别来区分。
气相色谱-质谱联用在法医毒物分析中十分重要,将分离效率极高的气相色谱仪与鉴定化合物可靠的手段质谱仪结合起来,二者都具有灵敏(可测到10-11克物质)、快速、可靠的优点,满足了毒物分析的各项要求。
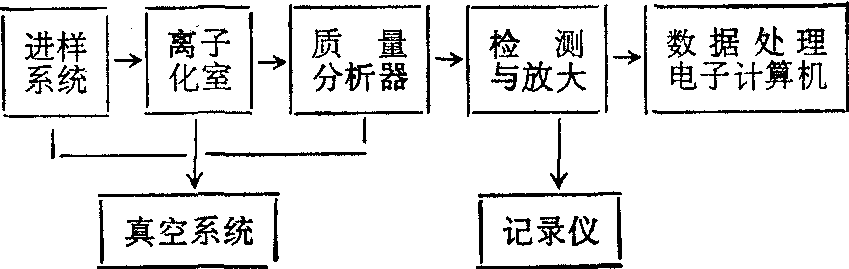
质谱仪基本结构示意图
在真空系统中,有机化合物在离子化室中经电子撞击电离(EI)或化学电离 (CI)等方法处理使之失去一个外层价电子而离解成为带正电荷的分子离子(molecular-ion,MH+)。MH+e-—→MH++2e-电子撞击电离
化学电离是先由载气如甲烷受电子撞击生成CH5+
CH4+e-—→CH4++2e-
CH4++CH4—→CH5++CH3然后CH5+与有机化合物分子反应生成带正电荷的分子离子MH+之外,尚生成一些碎片离子。这些分子离子和碎片离子,由于质量不同,通过质量分析器(静电分析器和磁分析器)分离,最后为离子捕集器收集,经放大记录,呈谱线形式显示,即为一化合物的质谱图。然后,以m/e(质量电荷比值)为横座标,以丰度为纵座标标绘质谱图。
m/e(质量/电荷)称质荷比,分子离子的质荷比相当于一化合物的分子量。
丰度,通常以最强的离子峰为基峰,其强度定为100%,计算其它峰的相对强度即丰度。
一些常见毒物的甲烷化学电离质谱数据
| 分子 量 | 药 物 | 分子离子(MH+) | 碎片离子 m/e>90 | |
| 质荷比 | 相对强度 | |||
| 135 135 137 138 155 162 164 165 178 | 乙酰苯胺 苯丙胺 异烟肼 水杨酸 美解眠 烟碱 水合氯醛 麻黄碱 可拉明 | 136 136 138 139 156 163 165 166 179 | (100) (100) (100) (100) (100) (100) (22) (29) (100) | 94(14) 119(65),91(9),134(5) 108(22) 121(90),95(8) 128(3) 161(27) 147(100),149(97) 111(75),113(49) 148(100), 164(8), 106(9),117(8) |
| 179 184 194 199 217 218 226 226 | 非那西丁 巴比妥 咖啡因 吩噻嗪 导眠能 眠尔通 异戊巴比妥 戊巴比妥 | 180 185 195 200 218 219 227 227 | (100) (100) (100) (100) (100) (5) (100) (80) | 152(11) 156(5),142(2),141(1) 138(2) 199(57) 190(9),189(6) 158(100),115(40) 157(12),156(11) 157(100),185(13), 156(8) |
| 231 | 氨基比林 | 232 | (100) | 231(40),113(20), 230(16) |
| 232 236 | 苯巴比妥 普鲁卡因 | 233 237 | (100) (12) | 204(6) 100(100),120(44), 164(9),235(5) |
| 238 | 速可眠 | 239 | (51) | 169(100),197(12), 168(11) |
| 242 | 硫喷妥 | 243 | (100) | 173(38),227(20) 187(10),201(10) |
| 247 250 252 255 | 度冷丁 安眠酮 苯妥英 苯海拉明 | 248 251 253 256 | (100) (100) (100) (52) | 246(20),174(5),202(3) 118(40),146(20) 175(25),210(7),225(4) 167(100),209(7), 254(5) |
| 266 | 安眠酮代谢 物 | 267 | (100) | 265(14) |
| 266 | 安眠酮代谢 物 | 267 | (100) | 118(6),134(4) |
| 270 | 安定代谢物 | 271 | (100) | 242(5),243(4) |
| 273 | 氯丙嗪代谢 物 | 274 | (78) | 273(100),238(59) |
(续上表)
| 分子 量 | 药 物 | 分子离子(MH+) | 碎片离子 m/e>90 | |
| 质荷比 | 相对强度 | |||
| 275 | 毒扁豆碱 | 276 | (100) | 275(75),219(55), 274(25),162(5) |
| 280 | 丙咪嗪 | 281 | (89) | 235(100),195(68), 208(59) |
| 284 | 丙嗪 | 285 | (100) | 284(96),212(12), 199(7),240(6) |
| 284 284 | 安定 异丙嗪 | 285 285 | (100) (100) | 162(6) 198(93),283(58), 284(54),240(34) |
| 285 289 | 吗啡 阿托品 | 286 290 | (32) (2) | 268(100),285(20) 124(100),281(1), 140(1) |
| 299 | 可待因 | 300 | (100) | 282(100),299(35), 298(21) |
| 299 | 利眠宁 | 300 | (100) | 299(38),264(24), 284(6) |
| 303 303 309 318 | 东莨菪碱 可卡因 美散痛 氯丙嗪 | 304 304 310 319 | (4) (20) (100) (100) | 138(100),156(6) 182(100),272(2) 265(60),308(28) 318(14),246(5), 248(4) |
| 324 | 奎宁 | 325 | (100) | 136(37),307(10), 323(7) |
| 334 352 | 士的宁 氟丙嗪 | 335 353 | (100) (72) | 334(30),33(22) 352(100),280(5), 282(4) |
毒物鉴定
identification of poison
- 锅塔豆腐是什么意思
- 锅塘是什么意思
- 锅墨是什么意思
- 锅墨灰是什么意思
- 锅墨烟是什么意思
- 锅大碗小是什么意思
- 锅头是什么意思
- 锅头渠儿是什么意思
- 锅头灶脑是什么意思
- 锅头的茄子——蔫的是什么意思
- 锅头饭好吃,过头话难说是什么意思
- 锅头饭好吃, 过头话难说。是什么意思
- 锅夹道是什么意思
- 锅套是什么意思
- 锅婆是什么意思
- 锅婆娘/子是什么意思
- 锅婆子是什么意思
- 锅嫌水壶黑——不知自丑是什么意思
- 锅子是什么意思
- 锅子菜是什么意思
- 锅小煮不烂牛头是什么意思
- 锅屋是什么意思
- 锅屑灰是什么意思
- 锅巴是什么意思
- 锅巴劫是什么意思
- 锅巴哪吒是什么意思
- 锅巴痞是什么意思
- 锅巴盐是什么意思
- 锅巴粥是什么意思
- 锅巴肉片是什么意思
- 锅巴蛋是什么意思
- 锅巴饭是什么意思
- 锅巴饭痂是什么意思
- 锅巴鱿鱼片是什么意思
- 锅巴黑勒是什么意思
- 锅巷是什么意思
- 锅师傅是什么意思
- 锅师父是什么意思
- 锅帘是什么意思
- 锅带子是什么意思
- 锅帽是什么意思
- 锅帽子是什么意思
- 锅庄是什么意思
- 锅庄舞是什么意思
- 锅底是什么意思
- 锅底上戳窟窿是什么意思
- 锅底上戳窟窿——捅漏子是什么意思
- 锅底下冒烟是什么意思
- 锅底云是什么意思
- 锅底坑儿是什么意思
- 锅底墨是什么意思
- 锅底屑是什么意思
- 锅底底是什么意思
- 锅底底儿是什么意思
- 锅底朝天是什么意思
- 锅底灰是什么意思
- 锅底的木头——烧焦了是什么意思
- 锅底的烟灰是什么意思
- 锅底脸是什么意思
- 锅底道是什么意思