遗传性酪氨酸代谢紊乱
遗传性酪氨酸代谢紊乱
遗传性酪氨酸代谢紊乱是一种先天性苯丙氨酸-酪氨酸代谢缺陷性疾病,属常染色体隐性遗传,由于对羟苯丙酮酸氧化酶及酪氨酸转氨酶缺乏或不足,致血中酪氨酸大量积聚并出现结节性肝硬化与肾小管损害。本病少见,散见于各地,特别发现于北欧斯堪的纳维亚与加拿大等地。
正常苯丙氨酸与酪氨酸代谢见图。苯丙氨酸第一步经苯丙氨羟化酶催化,不可逆转地变成酪氨酸,后经酪氨酸转氨酶与对羟苯丙酮酸氧化酶的作用生成尿黑酸,尿黑酸经尿黑酸氧化酶的进一步分解,最终生成延胡索酸与乙酰乙酸。
本病患者由于体内缺乏酪氨酸转氨酶或对羟苯丙酮酸氧化酶,致酪氨酸正常代谢过程中止,血中酪氨酸大量蓄积,并从尿液排出,其代谢底物如对羟丙酮酸、对羟苯乙酸与对羟苯乳酸亦从尿中大量排出。同时由于酪氨酸代谢障碍,影响蛋氨酸的正常分解,血中蛋氨酸浓度增加,约为正常人的10倍,病变后期酪氨酸代谢产物沉积于肾脏,致近端肾小管有广泛性损害,出现全氨基酸尿,并发生佝偻病。肝脏有结节性硬化,并有凝血障碍与程度不同的肝功能损害。由于酶缺陷的部位不同,有人将本病分为Ⅰ型与Ⅱ型,前者为对羟苯丙酮酸氧化酶缺乏引起,后者由酪氨酸转氨酶缺乏所致。
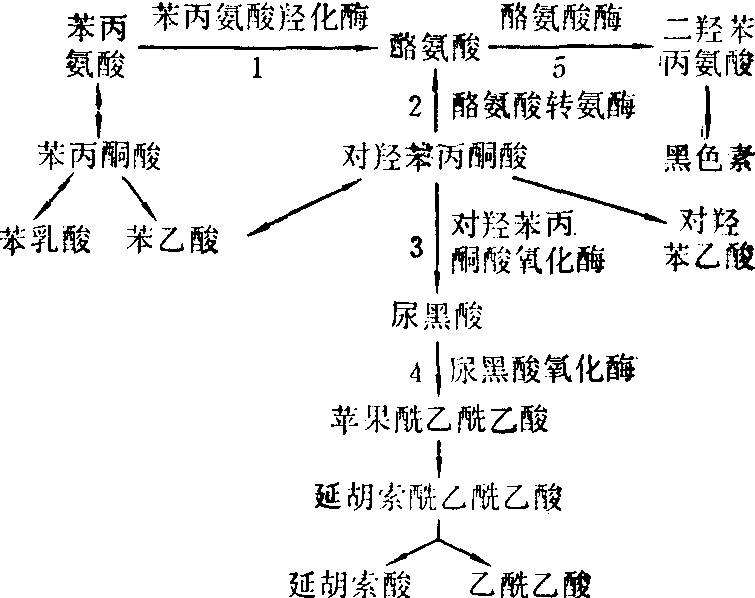
苯丙氨酸与酪氨酸代谢及代谢缺陷
1.苯丙酮尿症 2.Ⅱ型酪氨酸血症 3.Ⅰ型酪氨酸血症 4.黑酸尿症 5.白化病
Ⅰ型根据临床表现又分为急性和慢性两型。急性型患儿常在出生后一月出现症状,常见呕吐、黄疸与肝肿大,部分患儿可散发一种带甜味的特殊气味 (蛋氨酸分解产物),多数在一年内死于肝功能衰竭或上消化道出血。慢性型患者多见于儿童或少年,多有Fanconi综合征(发育障碍、肝肿大、佝偻病、糖尿及全氨基酸尿),部分出现神经精神症状。本病Ⅱ型很少见,主要表现为眼部症状(角膜溃疡、白内障)与皮肤损害(角化病、红斑)。
本病应尽早治疗,避免含苯丙氨酸与酪氨酸饮食,可使早期症状、眼睛与皮肤损害恢复,大量维生素D可望治愈佝偻病。
☚ 组织细胞增生病X 门静脉高压症 ☛
- 肚子里明是什么意思
- 肚子里有学问,不在穿的好坏是什么意思
- 肚子里有牢骚不敢喝酒是什么意思
- 肚子里有货是什么意思
- 肚子里没啥是什么意思
- 肚子里没墨水是什么意思
- 肚子里没有墨水是什么意思
- 肚子里没横肠——不懂得拐弯抹角是什么意思
- 肚子里烧起火,鼻子里也不要冒烟是什么意思
- 肚子里玩杂戏是什么意思
- 肚子里的萤火虫——心里明白是什么意思
- 肚子里盛不下二斤油是什么意思
- 肚子里编成是什么意思
- 肚子里能撑船是什么意思
- 肚子里蛔虫——知道心思是什么意思
- 肚子里装公章——心心相印是什么意思
- 肚子里装着二十五只小耗子——百爪挠心是什么意思
- 肚子里道道多是什么意思
- 肚子里长牙是什么意思
- 肚子里长草是什么意思
- 肚子里飞进萤火虫——全明白了是什么意思
- 肚子长出铁来——瘪不下去是什么意思
- 肚子难受是什么意思
- 肚子饿了是什么意思
- 肚子饿了填黄连是什么意思
- 肚孬是什么意思
- 肚孭是什么意思
- 肚实是什么意思
- 肚实实是什么意思
- 肚尖儿是什么意思
- 肚尾是什么意思
- 肚尾花是什么意思
- 肚屎是什么意思
- 肚屎下是什么意思
- 肚屙是什么意思
- 肚山是什么意思
- 肚巴脐儿是什么意思
- 肚帕是什么意思
- 肚带是什么意思
- 肚带绳是什么意思
- 肚底是什么意思
- 肚底货是什么意思
- 肚底黄是什么意思
- 肚当是什么意思
- 肚才是什么意思
- 肚拉是什么意思
- 肚拨羔是什么意思
- 肚捆是什么意思
- 肚排骨是什么意思
- 肚明是什么意思
- 肚末脐儿是什么意思
- 肚杂是什么意思
- 肚束三篾是什么意思
- 肚松是什么意思
- 肚板儿是什么意思
- 肚枵是什么意思
- 肚查是什么意思
- 肚档是什么意思
- 肚棓是什么意思
- 肚棚骨是什么意思