遗传性凝血因子缺乏
遗传性凝血因子缺乏所引起的出血性疾病比较少见。据国内统计,只占总的出血性疾病的4.5~16.8%。这类出血性疾病均按一定的遗传学规律通过基因从亲代遗传给后代。遗传性凝血因子缺乏症的遗传特点可分为以下几类:
(1)常染色体显性遗传: 如一般血管性血友病,异常纤维蛋白原血症,Passovoy因子缺乏症。
(2)常染色体隐性遗传:如因子Ⅴ、Ⅶ、Ⅹ、Ⅺ、Ⅻ、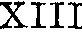 缺乏症,凝血酶原、纤维蛋白原缺乏症, 异常凝血酶原血症,Fitzgerald因子缺乏症,Fletcher因子缺乏症。
缺乏症,凝血酶原、纤维蛋白原缺乏症, 异常凝血酶原血症,Fitzgerald因子缺乏症,Fletcher因子缺乏症。
(3)Ⅹ染色体伴性隐性遗传: 如血友病甲、乙。
一般说来,遗传性凝血因子缺乏症只缺乏一种凝血因子,复合缺乏几种因子如血友病甲伴发因子Ⅴ、Ⅶ、Ⅺ或Ⅸ缺乏,伴发血管性血友病以及其他凝血因子缺乏者都很少见。在遗传性凝血因子缺乏症中,血友病甲和血友病乙最为多见,约占其总数的2/3以上,血管性血友病次之,其他少见。大多数遗传性凝血因子缺乏症都可有皮肤、粘膜、肌肉、关节、内脏等处出血,随凝血因子缺乏的程度而定。一般都需要作实验室检查才能确诊。大多数是凝血因子合成减少,少数可因分子结构异常导致凝血活性减低而致出血。目前尚无根治方法,都需要输全血、血浆或各种凝血因子浓缩制剂进行防治才能收效。
除血友病甲、乙,血管性血友病,因子Ⅺ缺乏症另有条目介绍外,以下叙述其他各种遗传性凝血因子缺乏症。
遗传性纤维蛋白原缺乏症 本症发现于1920年,极少见,是一种常染色体隐性遗传的出血性疾病。约有半数的病例有近亲婚姻史,纯合子患者有出血症状,杂合子患者可无出血史。
约有2/3的病人10岁以前即被发现有出血倾向。自发出血极少见,主要表现为创伤或手术后出血。关节出血较少见。伤口愈合可不佳。
凝血象检查示凝血时间、复钙时间、凝血酶原时间(PT)、活化或白陶土部分凝血活酶时间 (APTT或KPTT)、凝血酶时间(TT)均明显延长,严重者无血块形成。用化学或免疫学方法检测纤维蛋白原,血浆中的含量极微,甚至<5mg/dl。由于纤维蛋白原缺乏,故血沉很慢,血小板粘附性及血小板对低浓度ADP的聚集反应降低。
治疗方法主要是输血浆或纤维蛋白原制剂。后者的首次剂量为1g/10kg,以后每天150mg/10kg,使血浆纤维蛋白原水平到达100mg/dl。纤维蛋白原的半衰期为3~4天,故应每3~4天输注一次。
异常纤维蛋白原血症 本症于1958年由Imperato及Detteri首先发现,至今已发现的异常纤维蛋白原已不下30余种,以发现的地名命名,如异常纤维蛋白原Parma,Paris I,Baltimore,Zurich I,Detroit,Metz,LosAngeles,Oklahoma等。
大多数呈常染色体显性遗传。男女发病数大致相等。这是一种分子病,其缺陷性质是纤维蛋白肽分离障碍,有些缺陷的性质尚不明了。
临床表现为有轻度出血倾向,有的反复发生血栓和肺栓塞或伤口愈合障碍。有些患者无症状,仅在检查时才被发现。
实验室检查特点为PT及TT都有不同程度的延长,但因子Ⅴ、Ⅶ、Ⅹ、凝血酶原、纤维蛋白原定量均正常。血栓弹力图r延长,ma减低。
目前尚无根治疗法。需要时补充正常纤维蛋白原制剂。
遗传性凝血酶原缺乏症 本症发现于1947年,极少见。这是一种常染色体不完全隐性遗传的疾病。男女都可发病。初次发病常于儿童或青年期。凝血酶原降至正常的8~15%时才有出血症状,主要为皮肤、粘膜出血,可有深部肌肉、关节出血。常于小手术后出血。杂合子患者可无出血症状。
检验结果示PT延长不能被血清、吸附血浆或Russell蛇毒所纠正。APTT也可延长,TT正常。
用维生素K治疗无效。需要时输血浆或冻干血浆。手术前输血浆20ml/kg,每天二次,以后每天15~20ml/kg。也可用凝血酶原复合物,1u相当于1ml血浆所含的有关凝血因子。
遗传性异常凝血酶原血症 本症是凝血酶原分子结构异常所引起的出血性疾病。1968年首先被发现,呈常染色体隐性遗传。现知的异常凝血酶原至少有5种,称为凝血酶原Cardeza,Barcelon,San Juan,Brussels,Padua。
出血症状与分子结构异常的严重程度有关。最严重的病例出血症状类似轻型血友病,其凝血酶原的凝血活性只有正常的5%。
检验结果的特点为PT延长,但若用免疫学方法检测则结果正常,说明异常凝血酶原仍保持其正常抗原性。
遗传性因子Ⅴ缺乏症 本症亦称副血友病(parahem-ophilia),首先是由Owren于1944年发现的,这是一种少见的常染色体不完全隐性遗传的出血性疾病。男女都可发病。纯合子患者血浆中的因子Ⅴ含量只有正常的8~10%,杂合子者可达22~60%。只有纯合子者有出血症状。
患者大多于3岁后发病,表现为皮肤、粘膜出血,肌肉、关节出血少见。轻微创伤或小手术后出血多见。
实验室检查示PT延长,可被正常新鲜血浆或吸附血浆纠正,不被正常贮存血浆纠正。凝血活酶生成试验异常。
治疗方法是输新鲜血或血浆。输新鲜血浆15~25ml/kg可使因子Ⅴ的水平提高到15~30%。手术后第一天每12小时输一次。因子Ⅴ的半衰期为14~36小时(平均24小时),故以后每天输一次,至少10~14天。
遗传性因子Ⅶ缺乏症 本症是Alexander等于1951年首先发现的,当时称为“血清凝血酶原转变加速素”(SPCA)缺乏症。文献上所报道的病例至今已在70例以上, 国内也有报道。 这是一种常染色体隐性遗传的出血性疾病,男女都可发病。纯合子患者因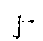 Ⅶ<1%;杂合子患者可达25~65%,无出血症状。血浆因子Ⅶ的水平<5%时才有出血。出血症状与血友病及凝血酶原缺乏症患者相似。关节可出血,但较血友病少见。近来发现本病患者易发生深静脉血栓形成及肺梗塞,机理不明。
Ⅶ<1%;杂合子患者可达25~65%,无出血症状。血浆因子Ⅶ的水平<5%时才有出血。出血症状与血友病及凝血酶原缺乏症患者相似。关节可出血,但较血友病少见。近来发现本病患者易发生深静脉血栓形成及肺梗塞,机理不明。
实验室检查示PT延长,能被贮存的正常血浆或正常血清所纠正,但不能被吸附血浆(不含有因子Ⅶ)纠正。
防治方法是输血或输凝血酶原复合物。因子Ⅶ的生物半衰期只有5小时,故必须每6小时输一次,每次5~10u/kg,可使因子Ⅶ维持在25%左右。
近年发现在有的病人因子Ⅶ的活性虽减低,但其抗原性仍正常,这种疾病是由于异常因子Ⅶ(abnormalfactor Ⅶ)引起的。
遗传性因子Ⅹ缺乏症 本症是Tilfer (1956年) 和Hongic (1957年)先后发现的。本病很少见,国内已有发现。这是一种常染色体隐性遗传的出血性疾病。男女都可发病。纯合子患者因子Ⅹ水平<1%,出血严重,新生儿期即可发病,表现为鼻衄、皮肤瘀斑、消化道出血、颅内出血。有的患者有关节、肌肉出血。杂合子患者因子Ⅹ水平可达50%左右,可无出血症状。
实验室检查示PT延长,能被贮存正常血浆或血清纠正,不被吸附血浆纠正,APTT延长。Russell蛇毒时间延长是确诊本病也是与因子Ⅶ缺乏症作鉴别的主要方法。
防治方法是输血、血浆或凝血酶原复合物。血浆的首次剂量为10~15ml/kg,以后每24~48小时输10ml/kg。
遗传性因子XII缺乏症 遗传性因子XII缺乏症或Hageman特性(Hageman trait)是Ratnoff及Colopy于1953年首先发现的,这是一种常染色体隐性或显性遗传的疾病。纯合子患者血浆中因子XII的水平可低至1%;杂合子者因子XII的水平可高达25~60%。
本病患者大多并无出血症状,甚至在手术时也不出血或出血甚微。少数有鼻衄或皮肤瘀斑。相反,因子XII缺乏症可伴发心肌梗死及血栓性静脉炎,说明并不能防止血栓栓塞性疾病的发生。
实验室检查示凝血时间及APTT均明显延长,凝血活酶生成障碍。须用已知因子XII缺乏症患者的血浆进行交叉试验,不能相互纠正者可确诊为本病。
因子XII明显减少而无出血症状的原因可能是通过因子XI激活内源凝血系统,例如血小板与血管内皮下组织中的胶原接触后可产生胶原诱导凝血活性(collage-n-induced coagulation activity),后者可激活因子XI,使血液循内源凝血系统进行凝固。
本病无需治疗。如有出血症状或需手术,输库血50~100ml即可奏效,效果可维持24小时。
Fletcher及Fitzgerald因子缺乏症 Fletcher因子缺乏症是由Hathaway等于1965年首先发现。是一种常染色体隐性遗传的特性(trait),现已弄清Fletcher因子就是前激肽释放酶(prekallikrein PK),PK在激活的因子XII(XIIa)作用下,转变为激肽释放酶(KallikreinK),PK转变为K时需要有表面接触(S)及高分子量激肽原(HMW-K)(见图)。K又可使高分子量激肽原转变为缓激肽(BK),也可进一步激活因子XII使成为XIIa,从而加速内源凝血系统的凝血。
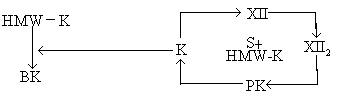
激肽释放酶及激肽系统的作用
Fitzgerald因子缺乏症是由Saito于1975年首先报道的,呈常染色体隐性遗传。现已证明Fitzgerald因子就是高分子量激肽原(HMW-K),它在内源凝血系统中的作用环节见上图。文献上所报道的Flaujeac因子、Williams因子都系激肽原,可能是同一物质。Fletcher及Fitzgerald因子缺乏时临床上均无明显出血,而APTT都延长,但不如因子XII缺乏症明显。这二种因子的缺乏症均无需治疗。
Passovoy因子缺乏症 本症是由Hongie于1975年首先报道,发生在一个家族4代5个成员中。这是一种常染色体显性遗传的疾病。临床上有些类似因子XI缺乏症,出血症状较轻,但手术后可出血不止。实验室检查也示APTT延长,可被正常血浆所纠正,不被正常吸附血浆所纠正,但因子IX正常。PT、TT、纤维蛋白原都正常。防治方法是输正常血浆补充此因子。
遗传性因子XIII缺乏症 本症是Duckert于1960年首先发现,很少见,至今文献上仅报道20余例,国内也有报道。这是一种常染色体隐性遗传的出血性疾病,男女都可发病。患者家族中常有近亲结婚史。纯合子患者血浆中因子XIII的水平<1%,杂合子者无出血症状。患者出生后不久即可被发现,但多数在成年期。临床特点是创伤或手术当时出血不多,但过12~36小时后出血变得明显。由于纤维蛋白的形成及原始纤维细胞增殖障碍,故伤口愈合延迟。少数患者有鼻衄、胃肠道出血、血尿、肌肉、关节出血、子宫出血等,有时易与血友病相混淆。常规凝血试验都正常,血栓弹力图ma减低。对本病具有特异意义的实验室发现是血块可在短时间内溶于5M尿素或1%单氯醋酸溶液(<1~2小时,正常24小时不溶解)。有的患者的血浆对抗因子XIII抗血清反应阳性,称为CRM+型,有的阴性称为CRM-型。
本病须与获得性因子XIII缺乏症相鉴别。获得性者较遗传性者多见,可发生于肝硬变、急性肝细胞坏死、淋巴瘤、多发性骨髓瘤、白血病、红斑狼疮、DIC、类风湿性关节炎、尿毒症以及结核病用异菸肼治疗时。
治疗方法是输全血或血浆,输血浆2~3ml/kg可使因子XIII的水平达到正常的5~10%,近期库血、纤维蛋白原制剂、冷沉淀物也有效。因子XIII的半衰期可长达150小时,故输血一次作用最长可维持4周,手术后每隔六天输注一次即可防止出血。
- 安娘是什么意思
- 安娜·卡列尼娜是什么意思
- 安娜·卡列尼娜 [俄国]列夫·托尔斯泰是什么意思
- 安娜·怀曼舞剧团是什么意思
- 安娜·林肯是什么意思
- 安娜·玛埃·帕克多是什么意思
- 安娜·路易斯·斯特朗是什么意思
- 安娜·阿赫玛托娃是什么意思
- 安娜·雅各布森·施瓦茨是什么意思
- 安娜尔·格丽是什么意思
- 安娜法令是什么意思
- 安娜贝尔·李是什么意思
- 安娥是什么意思
- 安娴是什么意思
- 安婆是什么意思
- 安婆沙洲是什么意思
- 安婶是什么意思
- 安媒是什么意思
- 安子介是什么意思
- 安子哇村是什么意思
- 安子文是什么意思
- 安子洼煤田是什么意思
- 安子瑛是什么意思
- 安宁是什么意思
- 安宁、和顺的样子是什么意思
- 安宁勿懈堕,有事不追遽。是什么意思
- 安宁区是什么意思
- 安宁县是什么意思
- 安宁县情是什么意思
- 安宁史是什么意思
- 安宁和平是什么意思
- 安宁和睦是什么意思
- 安宁太平是什么意思
- 安宁宣抚司是什么意思
- 安宁寂静是什么意思
- 安宁州是什么意思
- 安宁市(连然街道)是什么意思
- 安宁幸福是什么意思
- 安宁无懈情,有事不迫遽是什么意思
- 安宁桃花会是什么意思
- 安宁河是什么意思
- 安宁河地震带是什么意思
- 安宁河谷平原是什么意思
- 安宁清谧是什么意思
- 安宁清闲是什么意思
- 安宁渡口是什么意思
- 安宁温泉是什么意思
- 安宁温泉记是什么意思
- 安宁温泉风景区是什么意思
- 安宁的一觉是什么意思
- 安宁的乡村田园生活是什么意思
- 安宁的岁月是什么意思
- 安宁的日子是什么意思
- 安宁而快乐是什么意思
- 安宁道中即事是什么意思
- 安宁郡是什么意思
- 安宁颂 [英国]柯尔律治是什么意思
- 安宁饱暖即神仙是什么意思
- 安宅是什么意思
- 安宅正路是什么意思