血红蛋白病与遗传
血红蛋白病与遗传
血红蛋白病属于分子病,是由于血红蛋白的分子结构或合成速率的异常而引起的遗传病,前者称为异常血红蛋白综合征,后者称为海洋性贫血综合征。据世界卫生组织估计,全世界约有一亿多人携带血红蛋白病的基因,因此是一种比较常见的遗传病。
血红蛋白的分子结构 血红蛋白(Hb)是人体红细胞内主要的蛋白质。每个红细胞内大约有2亿8千万个血红蛋白分子。人体血红蛋白是一种球形的巨大分子,每个分子由近一万个碳、氢、氮、氧、硫原子和4个铁原子构成,分子量为66,700,体积为64×55×50A。血红蛋白是一种结合蛋白——色素蛋白,由珠蛋白和血红素构成。每一个珠蛋白分子有二对(4条)肽链,一对是α链,各由141个氨基酸残基构成; 另一对是非α链(β、γ、δ
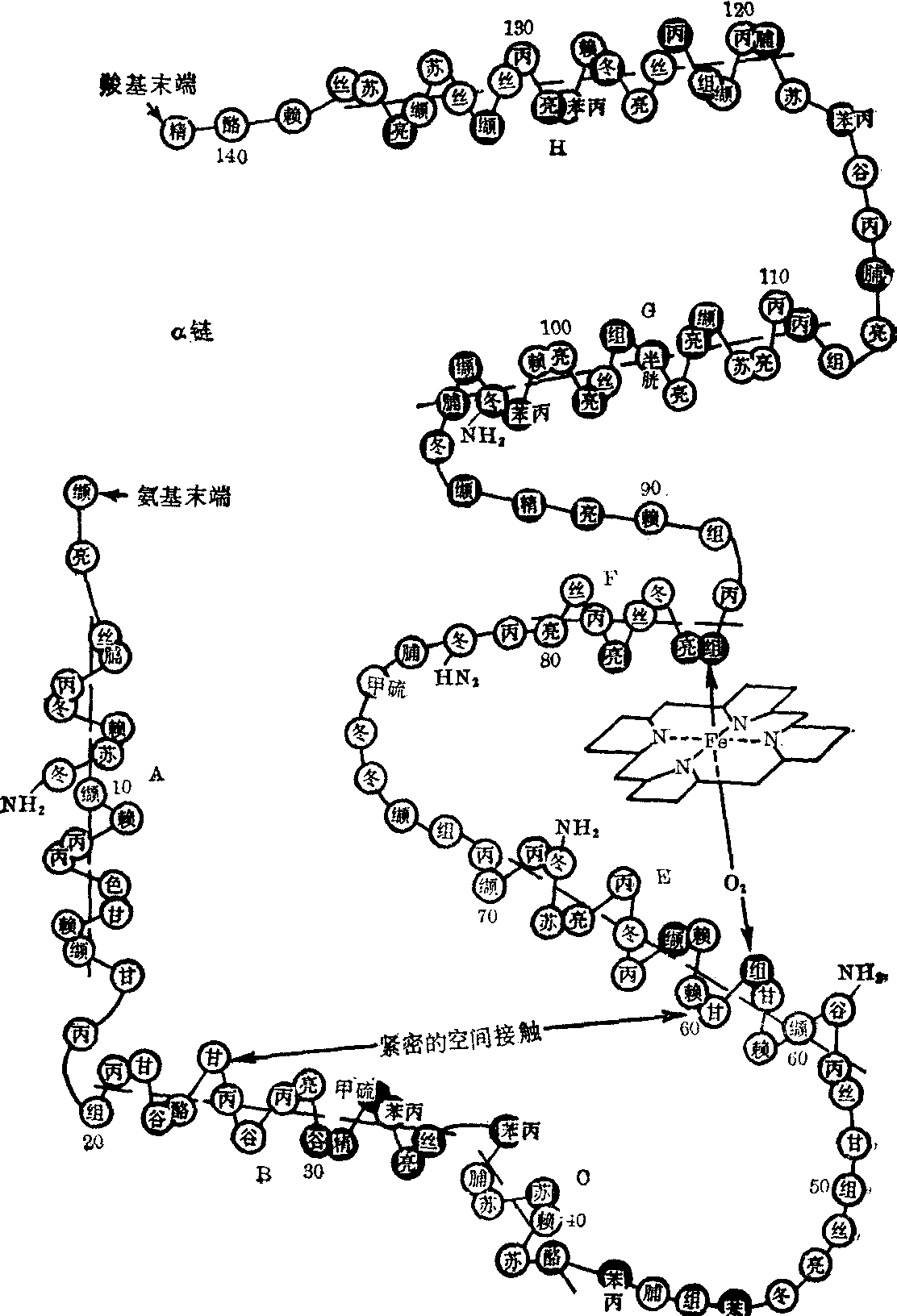
图1 血红蛋白α链
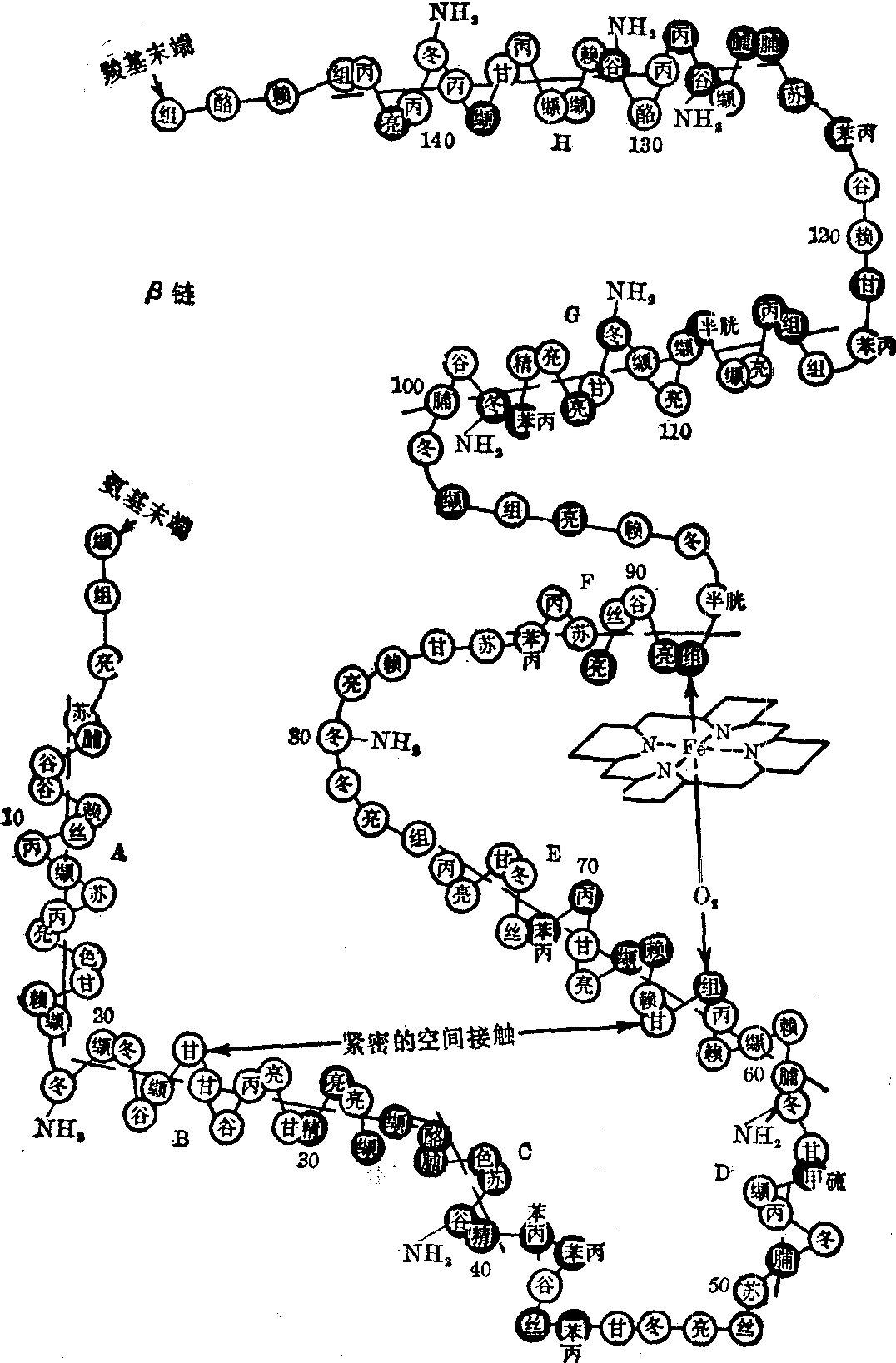
图2 血红蛋白β链
链),各由146个氨基酸残基构成。血红蛋白肽链的氨基酸排列顺序见图1,图2。每一条肽链和一个血红素连接,构成一个血红蛋白单体。人体血红蛋白分子就是由2对(4条)血红蛋白单体聚合而成的四聚体(图3)。
按照四聚体的肽链组成,人体的血红蛋白可以区分为下面三种:
❶血红蛋白A(HbA):α2β2, 是成人的主要血红蛋白成分,占95%以上;
❷血红蛋白A2(HbA2):α2δ2,是成人的次要血红蛋白成分,占2~3%;
❸血红蛋白F(HbF):α2γ2,称为胎儿血红蛋白,出生时占体内血红蛋白的70~80%,以后渐减,到生后6个月时,含量降低到1%左右。构成HbF的γ链有二种亚型: 一种是在γ链第136位上的氨基酸为甘氨酸,简称Gγ; 另一种是丙氨酸,简称Aγ。因此HbF相应也有二种,分别表示为α2Gγ2和α2Aγ2。
异常血红蛋白综合征 血红蛋白在人体内的主要功能是运送氧气,包括携带氧和释放氧两个方面,这和血红
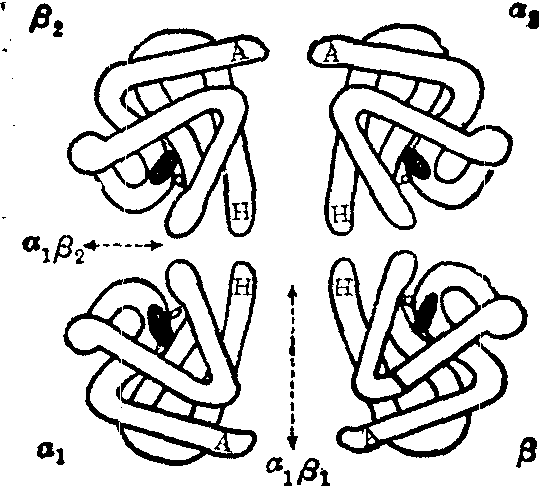
图3 血红蛋白分子结构
黑色扁平结构代表血红素,各和一条珠蛋白α链或β链结合,四个单体聚合成一个血红蛋白四聚体,即一个血红蛋白分子
异常血红蛋白的分子遗传学 血红蛋白的不同肽链是由各别的珠蛋白基因控制的,α基因位于16号染色体;β、δ、γ基因位于11号染色体,呈连锁关系。异常血红蛋白的产生是由于珠蛋白基因的DNA碱基发生变化,我们可以按碱基的变化和由此导致的异常血红蛋白归纳为五类:
(1) 单个碱基替代:产生单个氨基酸替代的异常血红蛋白。按照遗传密码的三联体假说,遗传密码上单个碱基替代,导致由它决定的氨基酸发生相应的变化,形成单个氨基酸置换的异常血红蛋白(图4)。在目前发现的异常血红蛋白中,绝大多数属于这种类型。
(2) 终止密码子突变: 导致肽链延长或缩短的异常血红蛋白。终止密码子的碱基发生变异,珠蛋白肽链的合成就不在正常的位置终止,而一直向C端延伸,到下一个终止密码子为止,结果导致肽链延长。相反,如果由于碱基的替代而形成异常的终止密码子,多肽链的合成就提 前结束,肽链因而就缩短(图5,图6)。
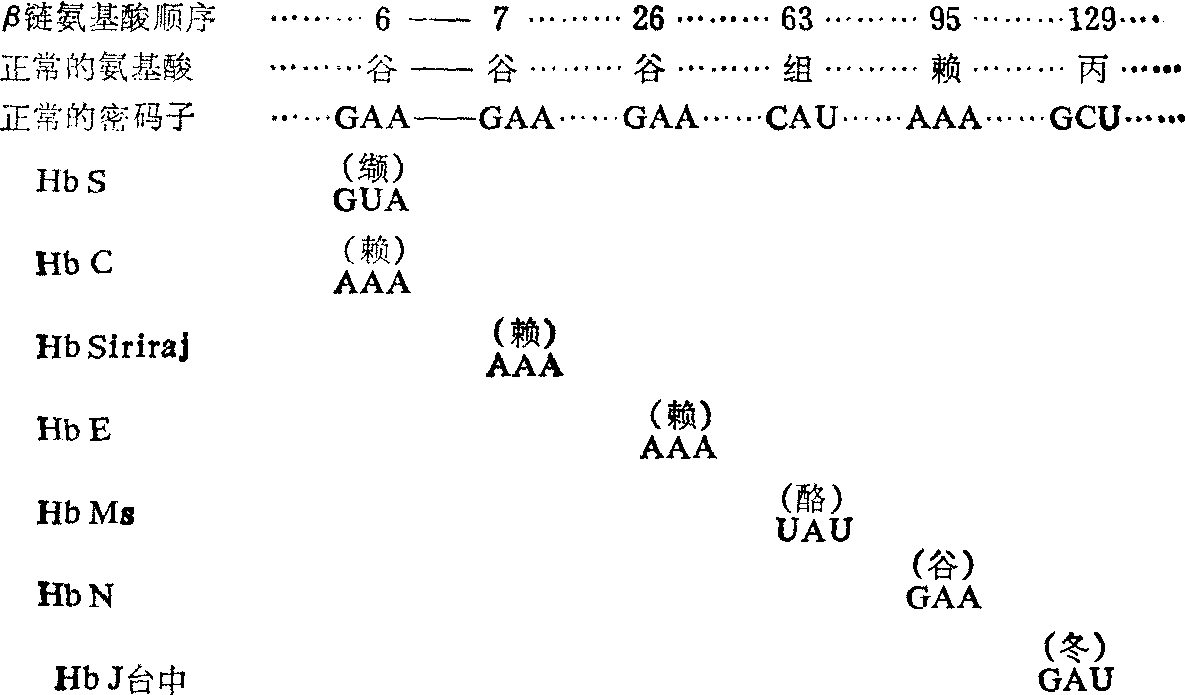
图4 正常和异常血红蛋白中氨基酸和密码子变化的比较

图5 人类α链mRNA终止密码区的碱基变异
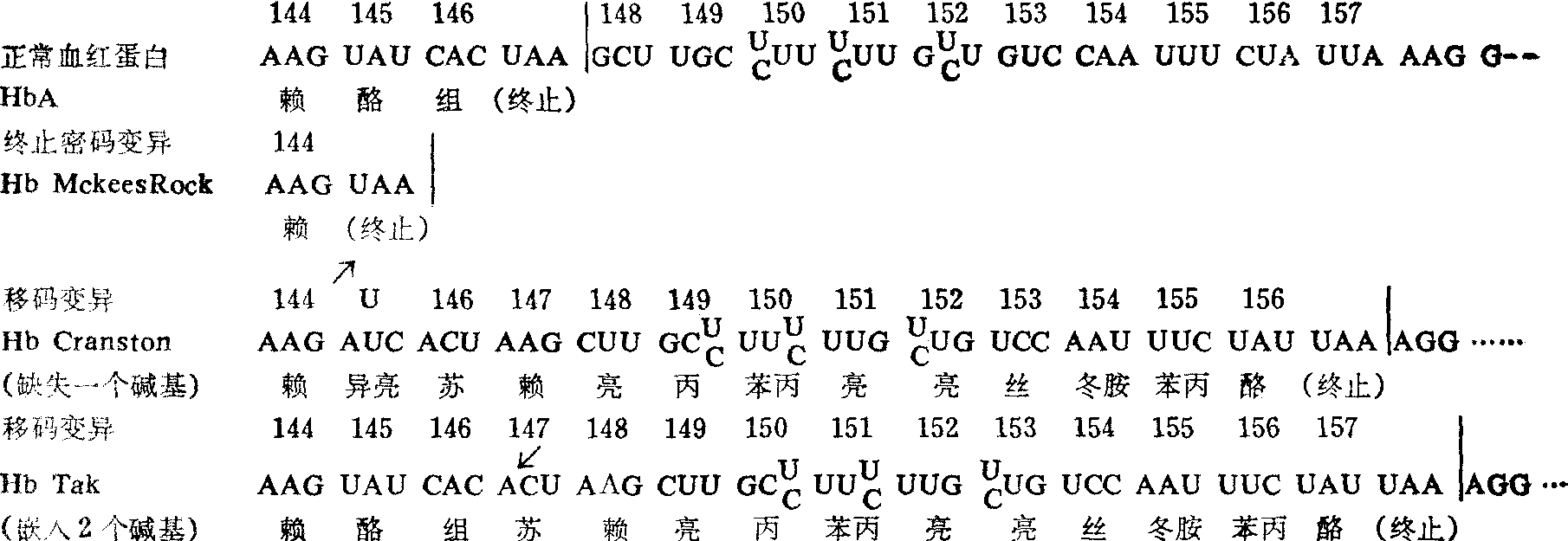
图6人类β链mRNA终止密码区的碱基变异
(3) 移码突变: 导致肽链氨基酸的长度和顺序发生改变的异常血红蛋白。如果在正常密码子中嵌入或缺失1~2个碱基,那么其后面的碱基便依次移后或推前。结果三联体密码子的碱基成分便发生变化。由此决定的氨基酸也发生改变。而且肽链的合成也将不在原来的位置终止,而在移码后出现的新的终止密码子处终止,致使合成的肽链增长或缩短。
(4) 密码子缺失和插入: 导致缺失或插入部分氨基酸的异常血红蛋白。由于在生殖细胞减数分裂时,联会中的染色单体发生错配和不等交换,结果形成两种珠蛋白基因,一种失去了一部分密码子,另一种插进相应的密码子,从而合成了缺失或插入部分氨基酸的肽链(表1)。
表1 缺失突变的异常血红蛋白
| 名 称 | 缺失的氨基酸 |
| Hb Leiden Hb Lyon Hb Freiburg Hb Niterai Hb Tochigi Hb St Antoine Hb Tours Hb Gun Hill Hb Leslie | β6或7谷 β17—18赖,缬 β22 缬 β42—44(或43—45)苯丙,甘,丝 β56—59甘,冬酰,脯,赖 β73—75甘,赖 β87 苏 β91—β96(或93—97)亮,组,半胱,冬 β131 谷 |
(5) 融合基因: 合成融合链的异常血红蛋白。由于减数分裂时不同珠蛋白基因之间发生不等交换,结果生成的珠蛋白基因含有原来两种不同珠蛋白基因的部分碱基,由此合成的肽链也就包含两种不同肽链的氨基酸。图7表示β和δ融合基因以及β和γ融合基因的形成过程。
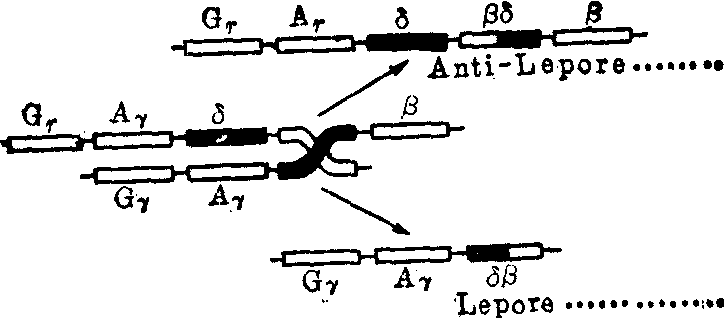
图7A δ和β基因错误联会后,发生不等交换,产生二种融合基因δβ(Lepore)和βδ(反Lepore)
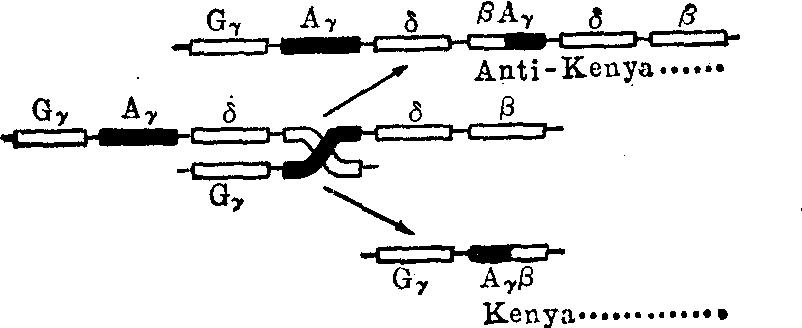
图7B Aγ和β基因错误联会后,发生不等交换,产生二种融合基因Aγβ(Kenya)和βAγ(反Kenya)
异常血红蛋白的分子病理学 异常血红蛋白病种类繁多,临床症状多种多样,但都可以从异常血红蛋白的结构变异所导致的功能变化来认识,按照结构变异的类型可以归纳为以下几种:
(1) 分子内部的氨基酸替代: 这类替代所产生的异常血红蛋白通常影响分子的构型和稳定性,导致功能的异常。这类异常血红蛋白包括血红蛋白M、不稳定的血红蛋白和氧亲和力改变的血红蛋白。
血红蛋白M(Hb M): 这种血红蛋白是肽链中与血红素铁原子连接的组氨酸或邻近的氨基酸发生了替代。最常见的是E7或F8的组氨酸为酪氨酸所替代。酪氨酸酚基上的氧与铁原子构成离子键,使铁原子呈稳定的高铁状态,因而影响了血红蛋白正常的运氧功能,导致组织供氧不足,出现紫绀症状和继发的红细胞增多,呈显性遗传,我国亦有发现。
不稳定血红蛋白(UHb): 肽链上与血红素紧密结合的氨基酸若发生替代,或者有关的氨基酸缺失,都会损害肽链的立体结构或减弱与血红素的结合力,形成不稳定血红蛋白分子。这种异常血红蛋白容易氧化,在红细胞内发生聚集沉淀,形成Heinz小体,改变细胞膜的硬度和功能,容易被脾脏破坏,引起溶血性贫血,因此又称为先天性Heinz小体溶血性贫血症(CHBHA)。这是比较常见的一类血红蛋白变型,迄今已发现80多个变型,国内也有一些发现。此外,在血红蛋白分子的α1β1接触面上的氨基酸若发生替代也会导致不稳定性。例如我国南方比较常见的血红蛋白E(Hb E) 就是这样一种异常血红蛋白,其β链第26位的谷氨酸为赖氨酸所替代,致使血红蛋白出现轻微的不稳定特性,纯合体红细胞渗透性降低,有轻度溶血性贫血。
氧亲和力改变的血红蛋白: 在羧基末端附近的氨基酸或α1β2接触面上的氨基酸发生替代,导致Hb的氧亲和力改变,已发现20多种。如果造成血红蛋白氧亲和力增高,输送给组织的氧不足,将促使肾脏分泌红细胞生成素,导致红细胞增多症;如果氧亲和力降低,则使动脉血的氧饱和度降低,严重者可引起临床上的紫绀症状。
(2) 分子外部的氨基酸替代: 已经发现一百多种在分子外部发生氨基酸替代的异常血红蛋白。一般来说,这类异常血红蛋白对分子构型、功能和稳定性都没有明显或直接的影响,个别异常分子的溶解度改变可导致临床症状。例如血红蛋白S(Hb S)是人体内发现的第一种异常血红蛋白,主要分布在非洲,在我国也有发现。分子结构的变化是β链上第6个氨基酸由谷氨酸变为缬氨酸(谷氨酸是带负电荷的极性亲水氨基酸,而缬氨酸是不带电荷的非极性疏水氨基酸),结果使Hb S溶解度下降5倍。Hb S纯合体体内Hb S含量多于95%,在氧张力低的毛细管区,Hb S分子间便由于溶解度下降而形成管状凝胶结构,使红细胞扭曲成镰刀状,这种僵硬的镰形红细胞不能通过微循环,加上凝胶化结构使血液粘滞性增加,阻塞微循环,引起局部缺血缺氧,产生痛性危象(如腹痛、关节痛等)。又由于这种镰形红细胞的细胞膜受到损害,被网状内皮细胞破坏,出现溶血性贫血症状,称为镰形细胞贫血症,患者多在成年以前死亡。Hb S杂合体的Hb S含量少于40%,一般并不出现严重的临床症状,但和β海洋性贫血组合成双重杂合体(βs/β thal
 )时,Hb S含量达60%以上,临床症状比杂合体严重得多。
)时,Hb S含量达60%以上,临床症状比杂合体严重得多。海洋性贫血综合征 海洋性贫血综合征是指血红蛋白的珠蛋白链合成速率降低所致的遗传病。全世界每年约有100万患儿死于本病。海洋性贫血综合征是一组具有多种遗传异常的疾病。按照受抑制的肽链而区分为α海洋性贫血、β海洋性贫血、δ海洋性贫血和δβ海洋性贫血等,重要的是α和β海洋性贫血。
(1) α海洋性贫血: α海洋性贫血是指α链的合成受到部分或完全抑制而产生的一组血红蛋白病。控制α链合成的基因(α基因)在16号染色体上,每条6号染色体原有二个α基因。这二个基因完全缺失,α链的合成便完全抑制,称为α海洋性贫血1(α-thal1);仅缺失一个α基因,导致α链合成部分受到抑制,称为α海洋性贫血2(α-thal2)。还有一种由于终止密码子突变产生的异常血红蛋白(Hb Constant Spring,HbCS),其合成α链的能力大大降低,后果和α-thal2相似。由上述三种基因相互作用构成α海洋性贫血的多种类型,其中α-thal1基因纯合体所致的Hb Bart’s水肿胎儿,以及α-thal1和α-thal2(或HbCS)基因双重杂合体所致的HbH病是二种重要
表2 α海洋性贫血类型
| α海洋性 贫血基因 | 纯合体 | 杂合体 | 双杂合体 | 分子遗传 基 础 |
| α-thal2 | 轻型海洋性 贫血,轻度 黄疸和血象 变化,出生 时有5~10 %的Hb Bart’s | 静止型海洋 性贫血,患 者无任何临 床症状,出 生时有1~ 2%Hb Bart’s | Hb H病,出 生时有25% Hb Bart’s 和微量Hb H,成人有5 ~30%Hb H和少量 Hb Bart’s, HbA2和Hb F含量降低 | 二个连锁的 α链基因中 有一个缺 失,因此转 录的mRN- A减少 |
| α-thal1 | Hb Bart’s 水肿胎儿, 有80%的 Hb Bart’s 以及少量的 Hb H和Hb Portland | 轻型海洋性 贫血,出生 时有5~ 10%的Hb Bart’s,但 HbA2和F 含量正常 | HbA2和F 含量降低, 出生时有 25%Hb Bart’和微 量Hb H | 二个连锁的 α链基因全 部缺失,以 致mRNA 缺失 |
| HLCS | 轻型海洋性 贫血,有5~ 6%HbCS | 静止型海洋 性贫血,出 生时有0.5 ~1%HbCS 和1~2% Hb Bart’s | 同上 | α链基因终 止密码子突 变(UAA→ CAA),导致 mRNA减 少 |
(2) 血红蛋白Bart's胎儿水肿综合征:在胎儿细胞两条16号染色体上共缺失4个α基因,因此完全不能合成α链,结果多余的γ链聚合成Hb Bart's (γ4),它对氧的亲和力高,造成组织缺氧,因此胎儿在子宫内发育到34~40周出现严重的全身水肿,死亡。最近已从Hb Bart's水肿胎儿的肝中分离出mRNA,这些mRNA能转译β链和γ链,但不能合成α链,证明胎儿缺乏α链的mRNA,进一步应用羊膜穿刺术,分离出Hb Bart's水肿胎儿细胞的DNA,并通过分子杂交,证实胎儿缺失α基因。
(3) 血红蛋白H病: 患儿是α-thal1和α-thal2或HbCS的双重杂合体,一共(或相当于)缺失3个α基因,由于α链合成减少,致使β链相对过多,β链合成HbH(β4)。HbH具有较高的氧亲和力,在正常生理条件下不能释放氧,失去血红蛋白正常的运氧功能。更重要的是HbH是一种不稳定血红蛋白,它含有较多的—SH基,氧化后导致β4离解为游离的β链。它在红细胞内沉淀积聚,附着于细胞膜上,形成红细胞的Heinz小体,致使红细胞膜受损,渗透性、脆性降低,易被脾脏的网状内皮系统破坏,病人出现脾肿大和溶血性贫血症状。
(4) β海洋性贫血:β海洋性贫血是指β链的合成受到部分或完全抑制而产生的一组血红蛋白病。如果β、δ链的合成同时受到抑制,则称为δβ海洋性贫血。一般用β°thal或δβ°thal表示β链或δ、β链合成受到完全抑制的基因,以β+thal或δβ+thal表示这些珠蛋白链合成受到部分抑制的基因。有一种由融合基因控制合成的血红蛋白Lepore,它的异常珠蛋白链是由部分δ链和部分β链联接而成,结果δ链和β链的合成速率均显著降低,表现和δβ°thal相似。各种β海洋性贫血基因相互合成海洋性贫血的多种多样的类型(表3)。
表3 β海洋性贫血类型
| β海洋性 贫血基因 | 纯合体 | 杂合体 | 分子遗传基础 |
| β+thal | 重型海洋性贫血,患者有严重的进行 性溶血性贫血,肝脾肿大和骨髓增生, 骨质疏松,常需输血治疗 | 轻型海洋性贫血,患者有轻度贫血、黄 疸和血象变比,HbA2含量升高 | β基因存在,但转录的βmRNA显 著缺乏 |
| β°thal | 重型海洋性贫血,症状同上,血中无 HbA,基本上为HbF | 轻型海洋性贫血,症状同上,HbA2含 量升高 | β基因存在,其缺陷可分为: ❶β mRNA缺失; ❷转录无功能的β mRNA; ❸βmRNA转译缺陷 |
| Hb Lepore | 重型海洋性贫血,症状同上,血中无 HbA和HbA2,含有75%HbF和 25%Hb Lepore | 轻型海洋性贫血,症状同上,血中Hb F含量升高,HbA降低,并有5~15% Hb Lepore | β-δ融合基因;βmRNA减少 |
| δβ°thal | 中间型海洋性贫血,症状介于重型和 轻型海洋性贫血之间,血中无HbA 和A2,100%为HbF | 轻型海洋性贫血,症状同上,有5~20 %HbF;HbA2的含量正常 | β基因缺失:δ基因可能缺失;β mRNA缺失 |
| δβ+thal | 未 报 道 | 轻度血液变化:HbF和HbA2含量 正常 | 基因存在 |
β海洋性贫血尚可和各种类型的海洋性贫血以及异常血红蛋白组成双重杂合体,构成复杂的临床综合征。例如βthal可以和Hb S、Hb E以及不稳定Hb复合,出现重型海洋性贫血症状,这些综合征在国内均有发现。
血红蛋白病的诊断 本病可根据临床症状和血红蛋白的合成异常在不同方面进行诊断。临床上有些有溶血性贫血症状(如海洋性贫血和不稳定血红蛋白病),有些有紫绀症状(如血红蛋白M病),有些根本没有任何临床症状。确诊须依赖实验室诊断。
本病可通过血液学检查,诊断是否存在由于血红蛋白合成速度降低导致低色素性红细胞特征,或者由于肽链合成不平衡而导致多余肽链在红细胞内沉淀,以及由于异常血红蛋白的不稳定性沉淀而产生Heinz小体,和由于Hb S而产生的红细胞镰状改变现象等。
在生化检验方面,对于异常血红蛋白综合征可通过电泳分析分离出异常血红蛋白成分,或者通过异丙醇试验和热不稳定试验鉴定不稳定血红蛋白,或者通过血红蛋白的溶解度测定,检出溶解度改变的异常血红蛋白。进一步还可通过分子杂交或肽链解离鉴定血红蛋白分子的异常肽链,并通过结构分析最终确定分子结构的变化。对于海洋性贫血综合征可通过测定血中Hb A2和Hb F的含量作实验诊断,进一步还可以通过测定血红蛋白的肽链mRNA含量来确定是否缺乏肽链mRNA,或者通过DNA分子杂交鉴定患者的肽链基因是否缺失。
血红蛋白病的治疗 目前尚无根治办法,但可根据本病特点,采取相应有效治疗措施。例如为了减少异常红细胞在脾脏中的破坏,对有严重脾功能亢进的病人可作切脾手术。对于有溶血性贫血的病人可输血改善症状,为了避免长期反复输血引起铁质沉着,可同时使用铁赘合剂(除铁胺)。从长远观点来看,既然血红蛋白病是由于血红蛋白肽链基因缺失或异常所致的疾病,因此可以通过基因工程技术矫正基因的缺陷。这是一项复杂的工作,但从目前的研究成果来看,血红蛋白病可能是最早由基因疗法治愈的疾病之一。
☚ 分子病 血浆蛋白病与遗传 ☛
- 城市地籍整理人员手册是什么意思
- 城市地籍管理是什么意思
- 城市垃圾是什么意思
- 城市垃圾的处理和利用是什么意思
- 城市垃圾资源化处理是什么意思
- 城市型集镇是什么意思
- 城市基准地价是什么意思
- 城市基准地价评估的方法和步骤是什么意思
- 城市基层社区服务是什么意思
- 城市基层社区管理结构是什么意思
- 城市基础设施是什么意思
- 城市基础设施建设是什么意思
- 城市基础设施的管理是什么意思
- 城市外围作战是什么意思
- 城市外围兵站是什么意思
- 城市外围防御是什么意思
- 城市外部联系是什么意思
- 城市外面的地方是什么意思
- 城市夜晚灯火通明是什么意思
- 城市夜景灯火通明是什么意思
- 城市夜景照明技术指南是什么意思
- 城市大气污染是什么意思
- 城市大气污染综合防治是什么意思
- 城市大气环境质量是什么意思
- 城市大酒店是什么意思
- 城市头是什么意思
- 城市媒介力是什么意思
- 城市学是什么意思
- 城市学校是什么意思
- 城市安置是什么意思
- 城市客流是什么意思
- 城市客流调查是什么意思
- 城市宫是什么意思
- 城市家庭是什么意思
- 城市家庭养老是什么意思
- 城市密布的地区是什么意思
- 城市对外交通是什么意思
- 城市对气候的影响是什么意思
- 城市对航线网是什么意思
- 城市导向标准应用指南是什么意思
- 城市导报是什么意思
- 城市小资产阶级是什么意思
- 城市就业结构是什么意思
- 城市居住区是什么意思
- 城市居住效率是什么意思
- 城市居民是什么意思
- 城市居民人均可支配收入是什么意思
- 城市居民人均居住面积是什么意思
- 城市居民人均消费水平是什么意思
- 城市居民储蓄存款余额是什么意思
- 城市居民地训练是什么意思
- 城市居民委员会是什么意思
- 城市居民委员会组织条例是什么意思
- 城市居民家庭全部收入是什么意思
- 城市居民家庭可支配收入是什么意思
- 城市居民最低生活保障是什么意思
- 城市居民最低生活保障制度是什么意思
- 城市居民消费品零售额是什么意思
- 城市居民消费心理是什么意思
- 城市居民生活质量指标是什么意思