真性红细胞增多症true polycythemia
系一种少见的,原因不明的慢性进行性造血系统疾病。本病晚期有变为白血病者,提示可能是一种红细胞系的肿瘤性疾病。亦可以骨髓纤维化而告终,因此有的将其归纳为骨髓增殖性症候群之一。由于红细胞过度增生导致血总容量增多和血粘度增加,使各脏器血管扩张、血流缓慢而出现的各种症状,如头痛、头胀、眩晕、耳鸣、皮肤与粘膜青紫、脾肿大、血管机能障碍、神经系统损伤及出血倾向等。治疗:可适当应用抗白血病药物、对症等。
真性红细胞增多症
真性红细胞增多症是一种原因不明的克隆性骨髓增生性疾病。据国外统计,每年的发病率为0.6~1.6/10万。犹太人发病较多。国内自1957年以来,据现有的材料至少有150余例。以31~60岁者居多,男性多于女性。本病的发病机理仍不清楚。过去认为是由于骨髓缺氧,有人设想是因为红细胞生存时间延长,还有人认为是红细胞生成素增加,但均经否定。近年来,有些学者报道真性红细胞增多症患者血清中可能有一种因子,刺激红细胞生成,刺激血红蛋白合成,对粒细胞生成也有轻微的刺激作用,称为骨髓刺激因子。此因子的抗原性与红细胞生成素不同,在体内需要红细胞生成素的参与才能起作用,它能促进小鼠干细胞池的生长、增殖,并加速多能干细胞向定向干细胞的转化,并使定向干细胞对红细胞生成素的作用更敏感。据认为此因子是糖蛋白,其特性尚需进一步研究。有人将Friend病毒滤液注射至Ha/ICR瑞士小鼠及DBA/2小鼠体内,可很快产生类似人类的真性红细胞增多症。由此考虑本病可能是病毒刺激红细胞增生。本病患者的红、粒及巨核细胞系均明显增生,染色体有异常核型,故又认为是多能干细胞疾病。本病与白血病的关系密切,有认为它们是同一病理过程的两种疾病; 也有认为白血病是真性红细胞增多症自然过程的不同病期,两者均为肿瘤性质。Dameshek将本病、白血病、出血性血小板增多症与骨髓纤维化症等总称为骨髓增生综合征。1970年Gilbert改称为骨髓发育异常综合征。
病理 主要的病理改变是长骨、扁平骨中的脂肪髓均被红髓替代,骨髓细胞增多,脂肪细胞明显减少,网状纤维稍增加,血窦高度扩张,窦内充满大量红细胞。脾脏轻度或中等度肿大,表面光滑,切面暗红,镜下见脾窦显著扩张,髓质增生及充血,滤泡萎缩,可见髓外化生。肝脏肿大,表面光滑,暗红色,镜下见肝窦扩张、淤血,可有髓外化生,也可有肝硬变的表现。此外可有血栓形成、出血、骨髓纤维化症等并发症的病理改变。
临床表现 起病缓慢,病人可无明显症状。有时可有疲乏无力及其他非特异表现。很多病人在偶然查血时发现; 也有的是经别人发现颜面发红检查血液发现。很多症状与血容量及血液粘滞性增加有关。患者感觉头晕、头胀、头痛、疲乏无力、耳鸣、视力模糊、怕热、出汗等。以后可有不同部位的静脉血栓形成,不同部位出血。胃或十二指肠溃疡发生率较正常人为高。国外报道全身发痒较多,国内并不多见。最多见的体征是皮肤及粘膜呈暗红色,两颊发红,四肢远端或末端呈紫红色。皮下可有出血点及紫斑,约半数以上患者有高血压,大部分病例有脾肿大,1/3~1/2有肝肿大。
实验室检查 有三方面的变化:
❶血象: 红细胞数大多在700~1,000万/μl; 血红蛋白18.0~22.0g/dl;红细胞压积60~80%。白细胞计数大部分高于正常,少数在正常范围内。粒细胞分类可有核左移现象; 个别病人可见中性中、晚幼粒细胞。血小板数亦常增多,有的病人正常;
❷骨髓象:大部分病人骨髓增生明显活跃或活跃,粒与红系比例大致正常。粒系以中性晚幼及杆状核粒细胞多见,嗜酸粒细胞稍多。巨核细胞多见,大多为有血小板形成的成熟型。个别病人骨髓增生减低;
❸其他化验:全身血总容量增加,红细胞容量明显增加,血浆容量可以正常、减少或增加。血液粘滞性比正常高5~8倍。血液比重达1.070~1.080。血沉缓慢,动脉血氧饱和度正常,中性粒细胞碱性磷酸酶高于正常,血清维生素B12结合蛋白、血液尿酸及组胺均增加。染色体可有非整倍体、假二倍体、多倍体、F组20号长臂缺失,最常见者为9号染色体的三体型。
诊断 典型病例具有皮肤及粘膜暗红色、脾脏肿大、全血细胞增多三方面的表现,同时伴有动脉血氧饱和度正常,诊断并不困难。国际真性红细胞增多症研究组制定的本病诊断标准为:
A.1. 红细胞容量(用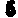 1Cr标记法)
1Cr标记法)
男性≥36ml/kg
女性≥32ml/kg。
2. 动脉血氧饱和度≥92ml。
3. 脾脏肿大。
B.1. 血小板数≥40万/μl。
2. 白细胞数≥12,000/μl(无发热或感染)。
3. 中性粒细胞碱性磷酸酶高于正常(>100)(无发热或感染)。
4. 血清维生素B12>900pg/ml。
未饱和维生素B12结合力>2,200 pg/ml。
凡符合A1+A2+A3或A1+A2再加B项中任何二项,则真性红细胞增多症的诊断可以成立。临床上,要注意与继发性和假性(应激性)红细胞增多症鉴别。
治疗 本病的治疗原则是: 静脉放血,使过多的红细胞与血容量恢复正常; 放射或化学治疗以抑制骨髓的造血功能,使红细胞、白细胞及血小板数控制在正常水平。两者亦可同时应用。
静脉放血 此法简便、易行、安全,在短期内即可使血容量恢复正常,症状消除。也可与其他治疗合用。静脉放血适用于有血管栓塞倾向者,在化学治疗或32P治疗前用以迅速减少血容量,外科手术前作为术前准备等。但放血后可发生继发性骨髓刺激现象; 多次放血可导致慢性缺铁;白细胞与血小板数增高者放血后常不能下降。具体方法是: 每次静脉放血300~500ml,隔2~3日一次,直至红细胞压积正常。老年人放血量宜适当减少,每次可放血250ml,每周一次。
放射治疗 以32P进行选择性内照射,32P集中在骨髓核分裂活跃的细胞中。常用的是Na2H32Po4,每平方米体表面积用2.3mCi,静脉注射,总量不超过5mCi。如在三个月内不见效,可再给药一次,较第一次用量增加25%,总量不超过7mCi。以后的治疗至少间隔6个月,全年总量不超过15mCi。口服量要比静脉注射用量增加25%,分次口服,每次2mCi,间隔7天。用药前给低磷饮食10天,服药前6小时及服药后3小时禁食,并继续低磷饮食半个月,以促进磷的摄取。用药后1~3月即有症状好转、脾脏缩小、血象好转。常可缓解几个月至一年以上。中数生存时间约为13年。其副作用主要是因剂量过大而发生骨髓抑制。关于32P治疗本病是否会诱发急性白血病,目前尚无统一意见。有人认为32P治疗者比非照射组急性白血病的发生率明显增高; 最近观察32P治疗组并不比瘤可宁治疗组的急性白血病发生率高。有人认为32P治疗后急性白血病的发生率与32P的剂量有关;但也有人认为32P治疗后,本病缓解期延长,生存时间亦延长,因而发生急性白血病的人数相应增多。其确切关系还未定。
化学治疗 适用于三系血细胞均增多,尤其是血小板明显增多者;有髓外造血,肝脾明显肿大者:有皮肤瘙痒、痛风性关节炎等并发症; 老年患者合并有心血管疾病不能放血者等。国内常用的为马利兰,也有用二溴甘露醇、瘤可宁、三尖杉酯碱等;国外还有用苯丙酸氮芥、环磷酰胺、阿糖胞苷、甲基苄肼、二溴丙基哌嗪等;
❶马利兰:每日2~6mg,分次口服。缓解时间可达一年左右。但可引起骨髓抑制;
❷瘤可宁: 每日6~8mg,分次口服。可以连续服用4周,再间歇4周,约有4/5病人见效。最近报道,治疗后发生急性白血病者较多;
❸苯丙酸氮芥(me-lphalan):开始每日6~10mg,5~7天后改为每日2~4mg,直至获满意效果后,改为每周2~6mg维持。有较好效果,副作用很少;
❹三尖杉酯碱: 高三尖杉酯碱或三尖杉酯碱每日2.5~5.0mg加于5%葡萄糖溶液500ml中静脉滴注,有一定效果,但副作用较多。
并发症的治疗: 本病如合并高尿酸血症、痛风性关节炎、尿酸肾病等,可用化学治疗,同时用别嘌呤醇,每日100~300mg,也可用秋水仙碱。全身皮肤痒用抗组胺药物,治疗效果都不明显,而化学治疗后常可见效。二苯环庚啶(Cyproheptadine)每日4~16mg,约对1/3病人有效;也有人用消胆胺(Cholestyramine)以控制严重皮肤瘙痒。晚期病人有贫血、且有脾大者,可输血,用雄激素;若有巨脾、脾机能亢进者,可行脾切除术。发展为骨髓纤维化症或急性白血病时,应按骨髓纤维化或白血病治疗。
手术治疗 选择性手术最好不做,以免出血不止或血栓形成。除非用化学治疗使血象恢复正常后再做。如需紧急手术,应先放血,直至红细胞压积正常时再手术。
病程及预后 本病发展缓慢,如无并发症,可存活10~20年。常见合并症有: 高血压、肝硬变、出血、血栓形成、胃及十二指肠溃疡、肾结石、胆结石、痛风性关节炎、骨髓纤维化症及急性白血病等。不治疗病例的中数生存时间仅18个月。经放血,化学治疗及(或)32P治疗的中数生存时间相似。主要死亡原因为血栓形成、出血、发生白血病或骨髓纤维化症。
- 济众水是什么意思
- 济伯是什么意思
- 济儒是什么意思
- 济光是什么意思
- 济克是什么意思
- 济克济札布是什么意思
- 济克美当木垂是什么意思
- 济兖公路是什么意思
- 济公是什么意思
- 济公传是什么意思
- 济公传说是什么意思
- 济公全传是什么意思
- 济公和尚的扇子是什么意思
- 济公岩是什么意思
- 济公的传说是什么意思
- 济公的圿圿——万应灵丹是什么意思
- 济公酒伴侣产品责任保险是什么意思
- 济农是什么意思
- 济农仓是什么意思
- 济利是什么意思
- 济利堂是什么意思
- 济办是什么意思
- 济助是什么意思
- 济勋是什么意思
- 济勒弥是什么意思
- 济勺是什么意思
- 济北是什么意思
- 济北先贤传是什么意思
- 济北国是什么意思
- 济北王兴居反逆案是什么意思
- 济北王宽禽兽行祝诅案是什么意思
- 济北王慕容泓是什么意思
- 济北诗人是什么意思
- 济北郡是什么意思
- 济南是什么意思
- 济南“救国十人团”是什么意思
- 济南一中是什么意思
- 济南七十二名士是什么意思
- 济南七十二名泉是什么意思
- 济南业余剧社是什么意思
- 济南东元盛染厂是什么意思
- 济南东站是什么意思
- 济南中豪大酒店是什么意思
- 济南义威桥是什么意思
- 济南之战是什么意思
- 济南习俗是什么意思
- 济南乡村师范学校是什么意思
- 济南书词公会是什么意思
- 济南书词娱化社是什么意思
- 济南书院是什么意思
- 济南二安是什么意思
- 济南五大行是什么意思
- 济南五峰山旅游区是什么意思
- 济南交通学校是什么意思
- 济南交通银行大厦是什么意思
- 济南亨得利表店是什么意思
- 济南人民出版社是什么意思
- 济南人民剧场是什么意思
- 济南人民广播电台是什么意思
- 济南人民曲艺队是什么意思